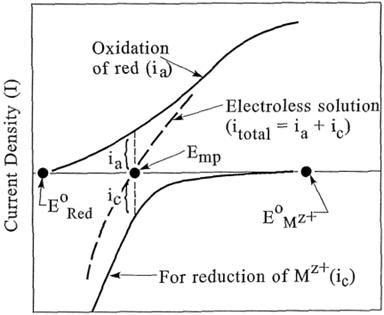
|
||||||||||||||||||||||||||

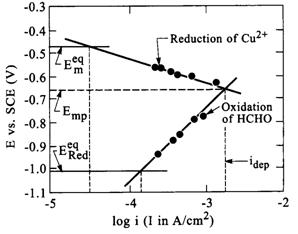 |
| Fig. 2. Current-potential curves for reduction of copper ions and oxidation of formaldehyde. �Eeqm� and �EeqRed� are the open circuit potentials for the copper ion reduction and formaldehyde oxidation reactions, respectively. The vertical lines represent the exchange current densities for the two half reactions, and the deposition current densityfor the complete electroless solution. (Paunovic, 1968) |
The MPT model was also reported to apply in a number other electroless metal deposition systems, including:
- electroless nickel from a citrate-complexant containing solution with dimethylamine borane (DMAB) reductant (Paunovic, 1983)
- electroless gold deposition from a gold cyanide containing solution, which utilized potassium borohydride as reducing agent (Okinaka,1973)
- more recently, cobalt-tungsten-phosphorus alloy (tungsten concentration not given, but probably was a few atomic %) from a solution that utilized sodium hypophosphite as reducing agent and ammonium tungstate as the source of tungsten ions (Shacham-Diamand and Sverdlov, 2000).
Though the simplicity and elegance of the MPT model have motivated much electrochemical type investigation of electroless systems, there are reports of solutions that do not operate in accord with MPT, including conflicting reports from different groups for electroless systems that appear on the surface to be similar. There are several possible reasons for this variability in observations between the various groups, including the following:
- the use of different metal ion complexants, which invariably leads to varying activities of free metal ions in solution
- different solution temperatures and pH;
- varying concentrations and types of additives used as solution stabilizers;
- the presence of varying concentrations of dissolved oxygen which undergoes a parallel reduction reaction;
- the use of different solution agitation conditions which has direct impact on the concentration of additive or dissolved oxygen at the surface;
- the use of somewhat different methods to obtain polarization curves (pseudo steady state against potential sweep method carried out at different sweep rates);
- use of chemicals of different grades of purity.
Catalytic aspects
The critical reaction in electroless deposition from the viewpoint of catalysis is oxidation of the reducing agent. The ability to catalyze oxidation of the reductant determines whether a metal or alloy can sustain electroless deposition in an otherwise properly formulated electroless solution.An interesting feature of the electroless deposition process is that no one metal appears to be a good catalyst for oxidation of all reducing agents that have been employed for electroless deposition. Thus, copper is an exceptional catalyst for formaldehyde oxidation, but is inactive for hypophosphite oxidation from a practical standpoint. Nickel and cobalt are poor catalysts for hydrogen oxidation, but are good catalysts for virtually every other reducing agent relevant to electroless deposition. Palladium appears to come closest as an example of a good catalyst for all reducing agents; however, it is likely that one could find a catalyst that could match, if not exceed, its activity in the case of each reductant. This, combined with its nobility, or resistance to dissolution in electroless solutions and to excessive oxidation, has made palladium the universal choice as a catalyst for initiating electroless deposition at catalytically-inactive metals and insulators.
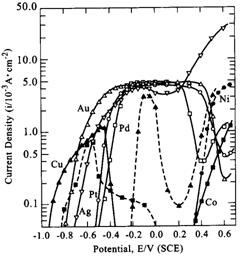 |
| Fig. 3. Current-potential curves for anodic oxidation of formaldehyde on various metals. Dotted lines: current densities attributable to the anodic dissolution of copper and cobalt electrodes. (Ohno, 1985) |
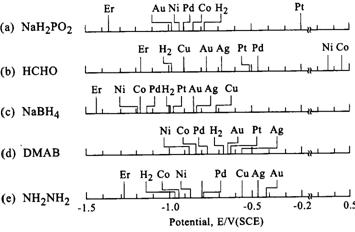 |
| Fig. 4. Catalytic activities of metals (as potentials measured at 10-4A/cm2 for anodic oxidation of reductants). �Er�: oxidation-reduction potentials of reductants. (Ohno, 1985) |
Similar polarization curve data has been obtained by Ohno (1985) for other important reducing agents. In an effort to classify the activity of various metal electrodes for the reducing agents, Ohno tabulated the potentials exhibited by the reducing agents for a fixed current density of
1.0 × 10-4 A/cm2; this data is tabulated in Figure 4 (catalytic activity of course increases on going from high to low potentials). Although the data in Figure 4 indicates trends that may be useful in designing electroless solutions, it refers to particular experimental conditions, and may not adequately describe catalytic activity behavior in different types of electroless solutions.
Mechanistic aspects
 Nickel-phosphorus
Nickel-phosphorus
The shapes of the polarization curves shown in Figure 3, including those generally observed for other reducing agents, are invariably complex, and not representative of simple electrochemical reactions. From the inception of modern electroless deposition practice, a number of mechanisms have been advanced to describe the electroless deposition process, many of which dealt with nickel-phosphorus. The classical mechanisms include the following:
- adsorbed atomic hydrogen was first postulated to be the reducing agent for nickel ion;
- hydride ions as the purported reducing species;
- metal hydroxide, in which hydrolyzed nickel ions were involved in mechanism of electroless deposition;
- An electrochemical mechanism, in which the deposition of nickel-phosphorus was an electrochemical process that occurred through local cells on the surface.
It is apparent from this review of electroless nickel-phosphorus deposition, by far the most important technological example of an electroless process with hypophosphite as reducing agent, that the mechanism for this reaction is still not fully understood. (See also the Appendix.)
Electroless copper
In contrast to typical solutions based on hypophosphite and dimethylamine borane reducing agents, which involve to co-deposition of phosphorus and boron, respectively, electroless copper tends to results in virtually 100% pure copper. Although electroplated copper has recently taken center stage as a replacement for aluminum interconnects in the back-end-of �line (BEOL) structures in ultralarge scale integrated (ULSI) technology, electroless copper has had much success in the past in a number of areas of microelectronics, for example, in generating the conductors in high-end glass epoxy circuit boards. In contrast to electroplated copper, which requires a global seed layer, electroless copper is readily selectively deposited on patterned, electrically isolated, circuit elements on suitably catalyzed (usually with dispersed, sub-colloidal palladium) dielectric surfaces.
Electroless copper has not been as widely investigated as the electroless nickel processes. Similarly, its mechanism does not appear to have been adequately described in the technical literature. Formaldehyde is still by far the most common reducing agent used to promote electroless copper deposition, usually in solution of pH = 10.5-12.5. Formaldehyde undergoes oxidation to formic acid or formate, depending on pH, a process that has a standard potential of +0.056 V (SHE); the standard potential for the copper redox couple is 0.34 V (SHE). See the Appendix for some proposed mechanisms.
Hydrogen evolution in electroless deposition
The co-evolution of hydrogen gas in most electroless deposition processes is an aspect of electroless deposition which needs to be understood, since it is integral part of the electroless deposition process for the majority of electroless solutions, and since it also impacts the properties of deposits by hydrogen inclusion.In a study of formaldehyde electrooxidation at various metals, Bindra and Tweedie (1985) provided insight into the oxidation of formaldehyde in the context of the differences of the energy of hydrogen adsorption on various metal catalyst surfaces. Dividing the latter into three categories, depending on their formaldehyde oxidation behavior they noted the following correlation:
- in the case of metals with positive energy of hydrogen adsorption, such as copper, formaldehyde oxidation is accompanied by hydrogen gas evolution;
- in the case of metals with energy of hydrogen adsorption close to zero, such as platinum and palladium, formaldehyde oxidation is not accompanied by hydrogen evolution;
- in the case of metals with negative energy of hydrogen adsorption, such as nickel, the kinetics of formaldehyde oxidation are extremely slow relative to the other two classes; these metals cannot be electrolessly deposited from solutions containing formaldehyde as a reducing agent.
As mentioned earlier, coevolution of hydrogen also occurs for other reducing agents employed in electroless solutions. A consequence of the involvement of adsorbed hydrogen intermediates in electroless deposition is that absorption of hydrogen occurs in the deposit, the extent of which depends on the deposit type (for example, less for copper than for palladium). If care is not taken in the case of palladium deposits, for example, films can deform and crack from stress minutes after removal from the electroless solution, due to rapid removal of the absorbed hydrogen from the metal. See the Appendix for mechanistic details.
Lacking the high-energy bombardment aspect of vacuum deposition, the electroless deposition process lacks a mechanism for deposits to adhere well to smooth insulator surfaces in the absence of special surface functionalization pretreatments. Compounding this problem is the stress caused by absorbed hydrogen. As solutions become more and more free of particles through constantly improving filtration methods, fewer particle-related nucleation sites, for example, hydrophobic organic material, exist for generating hydrogen gas bubbles in solution. Eventually, this can result in nucleation of hydrogen bubbles in �active� sites in the weak structural link of the insulator/deposit/solution system, namely the insulator/deposit interface, and film delamination may result.
Alloy deposition
Electroless deposition, with only a few exceptions, yields an alloy deposit, such as nickel-phosphorus. The present discussion principally deals with ternary and quaternary alloy deposits, for example, nickel-X-phosphorus in the case of electroless nickel-phosphorus-based alloys, where X is a third metal or element. Alloy deposition is thus an important feature of electroless deposition due to the ability to create new materials with interesting, and often unique, properties, such as good diffusion barrier, corrosion resistance, and magnetic properties. For a survey of the range of metals and elements which have been codeposited in electroless alloys, the reader is referred to Ohno (1998). Alloys containing mainly catalytically active metals
Magnetic recording has been a major driving force behind research into electroless cobalt alloys. Since this subject has been thoroughly reviewed elsewhere (Okinaka, 1994), attention in this section will focus primarily on a few recent examples illustrative of the alloy deposition reaction. Electroless alloys containing boron possess somewhat different physical properties to the analogous phosphorus containing alloys, and despite the higher cost of dimethylamine borane reducing agent relative to hypophosphite, they continue to receive attention.
In a recent study of nickel-cobalt alloy deposition using dimethylamine borane in the pH range 4-8, Saito (1998) observed that the rate of deposition of nickel-cobalt-boron increased with increasing nickel content of the alloy, which is consistent with higher catalytic activity on the part of nickel-boron for dimethylamine borane electrooxidation. Similarly, boron content increased with increasing nickel content, which is most likely due to the higher catalytic activity exhibited by nickel-boron for boron deposition, and not an energetic predisposition. Saito also noted that the boron content in the alloy tended to be inversely proportional to deposition rate. It was also noted that the partial current densities measured at the mixed potential values for cobalt-boron and nickel-boron solutions without reducing agent were much lower that the deposition rates determined by mass gain at electrolessly plating substrates. This indicated interaction between the partial anodic (reducing agent oxidation) and cathodic reactions (metal reduction, but also some phosphorus and boron reduction), and that a simple mixed potential model of electroless deposition (obtained by simply adding the anodic and cathodic partial reaction polarization curves) was incapable of describing the electroless solutions.
The incorporation of a third element, for example, copper, in electroless nickel-phosphorus coatings has been suggested to improve thermal stability and other properties of these coatings. Chassaing (1993) carried out an electrochemical study of electroless deposition of nickel-copper-phosphorus alloys (55-65% nickel, 25-35% copper, 7-10% phosphorus). As mentioned earlier, pure copper surfaces do not catalyze the oxidation of hypophosphite. They observed interactions between the anodic and cathodic processes: both reactions exhibited faster kinetics in the full electroless solutions than their respective half cell environments (mixed potential theory model is apparently inapplicable). The mechanism responsible for this enhancement has not been clearly established, however.
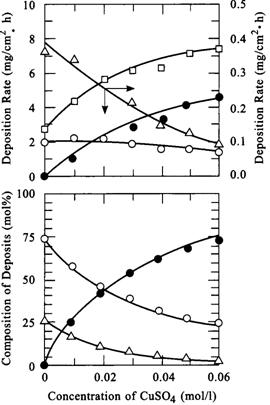 |
| Fig. 5. Effect of copper sulfate concentration (mol/l)on deposition of rate and composition of nickel-cobalt-phosphorus alloy. Squares: alloy; circles: nickel; filled circles: copper; triangles: phosphorus. (Nawafune, 1998) |
Clearly the rate of phosphorus deposition decreases along with the phosphorus content in the deposit, that is, it appears that it is not simply a case here of the sluggish phosphorus deposition reaction being dominated by the faster kinetics of the copper deposition reaction. Chassaing tentatively suggested that there existed an interaction between the phosphorus and copper deposition reactions. However, it may simply be the case that a small amount of nickel acts as a catalyst for hypophosphite oxidation, and the electrons derived from this reaction are conducted to sites where copper ions gets reduced to copper metal. The faster kinetics of copper deposition would then be largely due to the potential of the copper couple being considerably more noble than that of the nickel one. The difference in deposition kinetics are thus based on energetic factors in this instance, especially since the stability constants of the citrate-copper complexes do not exceed the stability constants of the citrate-nickel complexes by a large margin.
Gold continues to be an important metal in the microelectronics industry. It can be electrolessly deposited using, for example, a borohydride solution. Molenaar (1982) used more novel thiosulfate-based solutions and studied deposition of a range of gold-copper alloys from an alkaline solution containing formaldehyde reducing agent, gold cyanide, and copper ions complexed with ethylenediaminetetraacetic acid (EDTA). In an electroless type solution, due to virtually complete inhibition by cyanide of the catalytic oxidation of formaldehyde on gold, the stable gold cyanide complex did not undergo reduction. However, introducing copper into the solution enabled Molenaar to obtain alloy deposits with gold content ranging from 5 to 99%. In order to have a sustained electroless reaction, it was necessary to have a small amount of catalytically active copper present on the surface the deposit to enable oxidation of the formaldehyde. The gold-copper deposits contained homogeneous mixed crystals with a characteristic lattice constant for each composition, a linear relationship between lattice content and metal content being obtained.
Alloys containing catalytically inactive metals
For practical as well as fundamental reasons, there has been considerable interest in the deposition of alloys containing metals such as tungsten, molybdenum, and tin. In their pure forms, these metals do not catalyze the oxidation of the usual electroless reducing agents. Therefore, their mechanism of codeposition is intriguing, and developing an understanding of it should help to better understand the mechanism of electroless deposition as whole. Obvious questions in ternary and quaternary alloy deposition include the effect of the third- or fourth-element-containing-ions in solution on the rate of electroless deposition, as well as on the phosphorus and boron contents in the case of alloys based on nickel-phosphorus and nickel-boron.
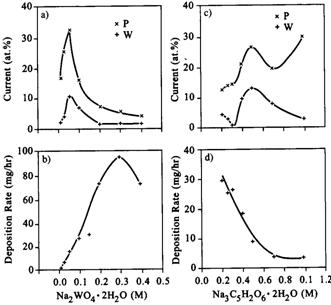 |
| Fig. 6. (a) and (b): Deposition rate and composition data for nickel-tungsten-phosphorus deposits as a function of sodium tungstate concentration. (c) and (d): Deposition rate and composition data for nickel-tungsten-phosphorus deposits as a function of sodium citrate concentration. (Bangwei, 1996) |
As summarized in O�Sullivan (2001), similar complex data has been reported for nickel-tin-phosphorus and nickel-tungsten-phosphorus alloy electroless deposition. Many of the studies in the literature concerning electroless alloys are difficult to comprehend from the viewpoint of deposition kinetics and mechanism. This is often due to the nature of the complexant used, which many times functions to complex not only the primary metal ion, such as nickel, but also the secondary metal, or element, ion. Thus, changing the concentration of the ligand in solution changes not only the concentration, or activity, of the free, or hexaquo, metal ion, but also that of the other species of ion undergoing deposition. Also, changing the concentration of the primary metal ion in solution alters the concentration of ligand available for complexing the secondary ion, also changing its free ion concentration, and so on. Having such interdependent concentrations of reacting ions in solution makes it difficult to extract the maximum possible information from fundamental studies of ternary alloy electroless solutions.
Electroless nickel-germanium-phosphorus was studied by this writer as a model system for ternary alloy deposition (O'Sullivan, 1993). A chloride-free solution with germanium oxide as a source of germanium, hypophosphite as reducing agent, aspartic acid as a selective complexant for nickel ions, which was operated at 80�C in the pH range of 5-5.8, was developed for depositing nickel-germanium-phosphorus films with a tunable germanium content from 0 to 25+ atomic %. The use of a complexant such as citric acid, which complexed germanium ions as well as nickel ions, resulted in a much lower germanium content in the electroless deposit, and a more complicated solution to study for the reasons discussed above. The aspartate-containing electroless solution, with its non-complexing pH buffer (succinic acid), approximated a �modular� system, and, with the exception of the aspartic acid-nickel complexation reaction, exhibited a minimum level of interactions in solution.
Other solution factors: additives and dissolved oxygen gas
Although electroless deposition seems to offer greater prospects for deposit thickness and composition uniformity than electrodeposition, the achievement of such uniformity is a challenge. An understanding of catalysis and mechanism, as discussed earlier, is often insufficient to describe the operation of a practical electroless solution. Solution factors, such as stabilizers, dissolved oxygen gas, and partially-diffusion-controlled, metal ion reduction reactions, often can strongly influence deposit uniformity. In the field of microelectronics, backend-of-line (BEOL) linewidths can be significantly less than 0.1 µm, which is much less than the diffusion layer thickness for a typical solution stabilizer additive, whose concentration may be 1 ppm (about 1-2 × 10-5 mol/dm3 lead). Good electroless deposit uniformity is expected to be difficult to achieve, especially in cases where a range of feature sizes is involved. As will be seen below, one cannot simply immerse a substrate with a nonuniformly distributed mask pattern into just any electroless deposition solution and expect to obtain uniform deposit thickness and composition. However, the situation can be significantly helped by good mask layout involving arrays of closely-spaced small circuit features, and avoiding isolated features, which may be subjected to non-planar diffusion of solution components. Solution additives
Strongly adsorbing solution stabilizers, for example, lead ions are often added to electroless solutions to prevent extraneous deposition. Such additives strongly impede the kinetics of reductant oxidation through an adsorption process: they are catalytic poisons. While suitable complexation of the metal ion(s) undergoing reduction reactions can yield a high degree of electroless solution stability, it is not always sufficient. This is because electroless deposition, being a catalytic process, can initiate at a variety of active sites in a complex deposition tool. The stabilizer additives adsorb on such sites, including on miscellaneous particles, and inhibit reductant oxidation, and hence deposition. Too high a concentration of additive at a catalytically active surface, brought about by either too high a concentration of additive in solution or as a consequence of hydrodynamics, can either prevent deposition from initiating, or can stop the deposition process.
Adsorbed additives also tend to undergo reduction during the electroless process, and become incorporated as impurities into deposits, most likely through a mechanism similar to that involved in ternary alloy deposition. In a manner similar to that discussed below in greater detail for dissolved oxygen, electroless deposition rates will be lower for features smaller than the stabilizer diffusion layer thickness. The edges of larger features, which experience higher stabilizer levels due to enhanced nonplanar diffusion, may experience reduced deposition rates, or may remain uncoated in extreme cases.
Dissolved oxygen gas
Oxygen gas is normally present in electroless solutions; unless steps are taken to reduce its concentration by inert gas purging, the concentration of dissolved oxygen usually approaches 10-5 mol/dm3 at room temperature. In contrast to deliberately added solution stabilizers, the oxygen molecule tends to adsorb weakly on catalytically active surfaces, where it may or may not undergo dissociation. Since at many metallic surfaces it is capable of being reduced via either a 2-electron or a 4-electron reaction route, the dissolved oxygen gas affects the mixed potential of the surface being coated in the electroless solution.
At most active electrocatalysts, including those of interest for electroless deposition, for example, palladium, the oxygen reduction reaction occurs at significant overpotentials under conditions of diffusion control. Thus, geometric effects related to size and distribution of plating features tend to be somewhat similar for both stabilizers and dissolved oxygen. For large-area substrates, the effect of dissolved oxygen on the kinetics of electroless deposition will generally be uniform throughout the substrate, except for edge regions. However, nonplanar diffusion effects on the kinetics of deposition need to be considered for feature sizes less than the oxygen diffusion layer thickness, the magnitude of which will be determined by hydrodynamics and temperature.
The oxygen reduction reaction affects not only the steady-state deposition kinetics, but also the initiation of deposition, the so-called induction time. At the beginning of the deposition process, the open circuit potential, �Eoc�, of either a uniformly catalytically active substrate, or a catalyst particle on an insulator, will be higher than that required for electroless deposition to occur. This is a consequence of the surface of the catalyst being covered with oxygen or hydroxide species which mask the catalytic activity of the surface; the value of �Eoc� would be expected to be in the range of +0.5 V to + 0.7 V (RHE) for a palladium surface. Normally, this is anodic, or positive, with respect to the �Em� value of the electroless reaction (Figure 1). Following removal of the oxide species from the catalyst surface, whether deposition subsequently initiates or not depends on the interplay between the kinetics of the parallel metal ion and oxygen reduction reactions, and oxidation of the reducing agent. Once an appropriate �Em� value is reached, metal deposition will occur.
In the absence of well laid out mask design, the presence of solution stabilizers and dissolved oxygen gas may impart a practical lower limit to the feature size that can be reproducibly fabricated using a particular electroless deposition; solution agitation will play a major role in determining this practical feature-size limit. Optimized electroless plating solutions which contain stabilizers to minimize particle generation and improve metallurgical properties, yet also contain dissolved oxygen, may not always be the best electroless solutions to employ in deposition in areas such as nanotechnology, unless the nano-sized features are uniformly laid out with close spacing in arrays, ideally with �fill� structures surrounding the arrays.
Some practical applications
 |
 |
| Fig. 7. (left) Complex-shaped structure which would be impossible to uniformly coat with conventional electrodeposition methods. (right) Generic computer disk drive. | |
In printed-circuit board manufacturing, electroless nickel phosphorus is commonly used as a coating with an overlay of gold to prevent corrosion, for example for connectors; the gold is put down by a self-limiting, chemical-displacement process. Electroless nickel is also used extensively in the manufacture of hard disk drives (Figure 7-right), as a magnetically neutral, smooth base coating on aluminum-based substrates (disks) prior to application of the magnetic read/write coating, which is usually done by physical vapor deposition (�sputtering�), and finishing with protective carbon and lubrication layers.
Electroless copper development underwent a quantum leap as a result of its use in printed wiring board (PWB) fabrication, where it is often referred to as additive technology. It was used for plating fine-line conductors and high-aspect-ratio through-holes, which ran from top to bottom of the multilayer circuit board, because of its high �throwing power� (the ability of a plating solution to cover uniformly) compared to the then-available copper electroplating processes. In the 1970s, IBM used this technology for the manufacture of multilayer circuit boards (MLBs) to package multichip modules, on which were mounted the logic chips, in its then top-of-the-line mainframe computers. This use of electroless copper led to much research and optimization of the process within and outside IBM, especially in the area of bath stability and the surface catalyzation using palladium colloidal-type �solutions�.
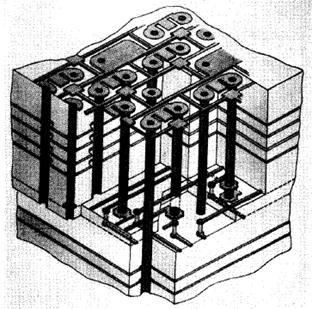 |
| Fig. 8. Isometric view of a printed-circuit board showing a portion of the thickness, internal signal interconnection, plated through-holes (heavy vertical lines) and details of surface interconnection pads for engineering change by wire bonding. (Seraphim, 1982) |
Nanostructured materials, such as nanoparticles, nanowires and nanoarrays, have attracted much attention due to their interesting electronic, optical and chemical properties. Electroless deposition has shown initial promise for fabricating nanostructures in this emerging field of research. This is not surprising, since an electrical connection is not needed to be set up to a nano-feature, as would be the case with electrodeposition (for example, using a conductive seedlayer), it being necessary to only set up the appropriate surface and electroless solution conditions for localized electroless deposition to proceed. Thus, several publications have appeared describing electroless deposition in nanopores, for example.
 |
 |
| Fig. 9. Scanning electron micrographs showing intersection (left) and network (right) structures of self-assembled palladium nanowires. (Shi, 2006) | |
Conclusions and future of electroless deposition
The mechanism of electroless deposition remains unclear. Given the complexity and variety of electroless deposition solutions available for study, this is not surprising. Another reason is that electroless deposition reactions have, for the most part, not been studied with the same rigor and intensity as have the hydrogen, oxygen, and methanol electrode reactions. In part this is due to the electroless deposition processes in the main working adequately well, generally in a short-term batch manner. The fuel cell related reactions have always held out the promise of bountiful energy, but have not yet delivered on those promises, hence the sustained study of such electrode reactions.Continuing interest in electroless deposition for microelectronics, which is relentlessly morphing into nanoelectronics, is generating fresh thinking on how best to apply electroless deposition to extremely small, and closely-spaced features (below 0.1 µm). The traditional methods of catalyzing the copper surface with palladium catalyst to initiate deposition invariably result in unacceptable copper surface roughness, and difficulties with completely removing the catalyst from the inter layer dielectric (ILD) regions, results in unacceptable levels of shorts. Also, traditional electroless solutions with low-concentration additives appear to be unsuitable for future microelectronic applications due to related deposit nonuniformity issues.
Initial efforts to generate short-lived electroless solutions at point of use, thereby avoiding the nonuniformity effects of additives, and to obtain electroless deposits without the use of palladium catalyst, have yielded encouraging results in the case of the self-aligned metal capping layer cobalt-tungsten-phosphorus and on copper interconnects, for example, in tests aimed at high performance logic chips at the 45 nm node and below. The main motivation for using these capping layers is to increase electromigration lifetime, with improvements in electromigration lifetime of over 100 times having been reported for the cobalt-tungsten-phosphorus cap. Avoidance of a catalyst (palladium) for initiating electroless hypophosphite reducing agent-based electroless solution is made possible by heating the solutions to a high enough temperature immediately prior to contact with the wafer, such that the slow kinetics of oxidation of hypophosphite at the copper interconnect surface, a poor catalyst for this reaction, are overcome.
As with other processes reliant on chemicals, electroless deposition will be forced to adopt more environmentally-friendly chemicals, for example, replacing the formaldehyde reducing agent in the case of electroless copper.
Appendix
The basic electroless deposition process
The basic electroless deposition process may be outlined with the following reactions occurring on the catalytic surface:
[1] ![]() Reductant ==> Oxidation product + ze-
Reductant ==> Oxidation product + ze-
[2] ![]() Mz+ + ze- ==>
Metal on substrate
Mz+ + ze- ==>
Metal on substrate
The overall electroless reaction may be depicted as:
[3] ![]() Reductant + Mz+ ==> Oxidation product + Metal on substrate
Reductant + Mz+ ==> Oxidation product + Metal on substrate
The failure of the proposed mechanisms, and related variations, to provide a comprehensive picture of electroless deposition involving hypophosphite reducing agent has sustained continuing investigations of the mechanism of electroless deposition. Observations such as co-evolution of hydrogen gas known to be derived from the reductant in electroless deposition, that the electroless process only occurs on metals generally known to be good hydrogenation-dehydrogenation catalysts, that sulfur-containing poisons for the latter class of catalysts act as electroless solution stabilizers, and that electroless deposition rates more often than not increase with increase in solution pH, prompted van den Meerakker (1981) to postulate a general mechanism that involves reductant adsorption followed by dissociation. This may be outlined as follows:
![]() Mechanistic aspects of the nickel-phosphorus deposition
Mechanistic aspects of the nickel-phosphorus deposition
[4] ![]() RH ==> RHads
RH ==> RHads
[5] ![]() RHads ==> R.ads + H.ads
RHads ==> R.ads + H.ads
[6] ![]() R.ads + OH- ==> ROH + e-
R.ads + OH- ==> ROH + e-
[7] ![]() H.ads + H.ads ==> H2
H.ads + H.ads ==> H2
Here, �RH� denotes the reducing agent, which yields the adsorbed radical species �R.ads� and atomic hydrogen upon adsorption and dissociation; the electron derived from the oxidation step (Equation [6]) goes towards metal ion reduction.
Van den Meerakker�s mechanism as embodied in Equations [4-7] is shown here as a possible general mechanism for reducing agent oxidation in electroless deposition. This writer does not mean to imply that this is the �actual� mechanism of reducing agent oxidation, or that it applies to all surfaces under all deposition conditions. Rather, the mechanism appears to be a good starting point for the study of a mechanism in a particular metal-reducing agent system.
Though equations such as [4-7] may be considered to represent the core steps, or reactions, of the reductant oxidation process in conventional electroless deposition, one or more additional reactions often need to be considered. In the case of reductants such as hypophosphite and dimethylamine borane, codeposition of phosphorus and boron, respectively, also occurs, as shown here for hypophosphite in acidic solution:
[8] ![]() H2PO-2ads + 2H3O+ + e- ==> 4H2O + P-B film on substrate
H2PO-2ads + 2H3O+ + e- ==> 4H2O + P-B film on substrate
Many factors influence the deposition kinetics of phosphorus and boron, including metal ion complexant concentration, solution pH, and temperature. Though they are unavoidable side products of the electroless deposits, phosphorus and boron impart unique properties to electroless deposits, for example, good corrosion resistance in the case of nickel-phosphorus deposits, where the phosphorus content can reach, or even exceed, 30 atomic % in certain solutions.
It is apparent from a review of electroless nickel-phosphorus deposition, by far the most important technological example of an electroless process with hypophosphite as reducing agent, that the mechanism for this reaction is still not fully understood. In the case of many of the solutions studied, it seems that a simple mixed potential model does not generally apply, which is not surprising when one sees, for example, the complexity of the steps in reactions [4-7], specifically the involvement of chemical steps, one of which is likely rate limiting.
Formaldehyde is still by far the most common reducing agent used to promote electroless copper deposition, In electroless solutions, formaldehyde undergoes transformation to a methylene glycolate anion, which is the actual reducing agent in electroless deposition:
![]() Mechanistic aspects of the electroless copper deposition
Mechanistic aspects of the electroless copper deposition
[9] ![]() H2CO + H2O ==> H2C(OH)2
H2CO + H2O ==> H2C(OH)2 ![]() (methylene glycol is a weak acid)
(methylene glycol is a weak acid)
[10] ![]() H2C(OH)2 + OH- ==> H2C(OH)O- + H2O
H2C(OH)2 + OH- ==> H2C(OH)O- + H2O
On copper, the overall anodic reaction involving formaldehyde, or more correctly, the methylene glycolate anion may be depicted as:
[11]
It is seen that one electron is obtained for each methylene glycolate anion on copper catalysts, although there is a possibility of obtaining two electrons if protons (or water in alkaline solution) were produced instead of hydrogen gas; the latter case tends to be observed at palladium and platinum catalysts (van Den Meerakker, 1981; Ohno, 1985; Bindra and Tweedie, 1985):
[12]
Thus, formaldehyde is not an efficient reductant in electroless copper solutions, not that this is an issue, given its continuous depletion by the parasitic homogeneous Cannizarro reaction. Van den Meerakker (1981) proposed a mechanism for methylene glycolate oxidation at copper which included a step involving dehydrogenation of adsorbed methylene glycolate anion:
[13]
[14]
[15]
[16]
Van den Meerakker�s mechanism is to some extent a �milestone� mechanism for electroless copper deposition that many researchers pay attention to, either to support, or, find fault.
[17]
If care is not taken in the case of palladium deposits, for example, films can deform and crack from stress minutes after removal from the electroless solution, due to rapid removal of the absorbed hydrogen from the metal. This is a consequence of the well-known ability of palladium, including electroless palladium deposits, to absorb hydrogen to a level approaching a palladium to hydrogen ratio of 1:0.8 or greater, the high relatively high diffusion coefficient of hydrogen in palladium, and the relatively fast kinetics of the recombination reaction (Equation [16]) occurring at the palladium surface.
Listings of electrochemistry books, review chapters, proceedings volumes, and full text of some historical publications are also available in the Electrochemistry Science and Technology Information Resource (ESTIR). (http://knowledge.electrochem.org/estir/)
Return to:
Top –
Encyclopedia Home Page –
Table of Contents –
Author Index –
Subject Index –
Search –
Dictionary –
ESTIR Home Page –
ECS Home Page
![]() H2C(OH)O- + OH- ==> HCOO- + H2O + 0.5H
H2C(OH)O- + OH- ==> HCOO- + H2O + 0.5H![]() H2C(OH)O- + 2OH- ==> HCOO- + 2H2O + 2e-
H2C(OH)O- + 2OH- ==> HCOO- + 2H2O + 2e-
![]() H2C(OH)O- ==> H2C(OH)O-ads
H2C(OH)O- ==> H2C(OH)O-ads
![]() H2C(OH)O-ads ==> HC(OH)O-ads + H.ads
H2C(OH)O-ads ==> HC(OH)O-ads + H.ads ![]() (rate determining step)
(rate determining step)
![]() HC(OH)O-ads + OH- ==> HCOO- + H2O + e-
HC(OH)O-ads + OH- ==> HCOO- + H2O + e-
![]() H.ads + H.ads ==> H2
H.ads + H.ads ==> H2
Hydrogen can absorb into the metal as:
![]() Mechanistic aspects of hydrogen absorption
Mechanistic aspects of hydrogen absorption![]() Hads ==> Hbulk
Hads ==> Hbulk
Related article
Electroplating
Bibliography
ECS | Redcat Blog | ECS Digital Library