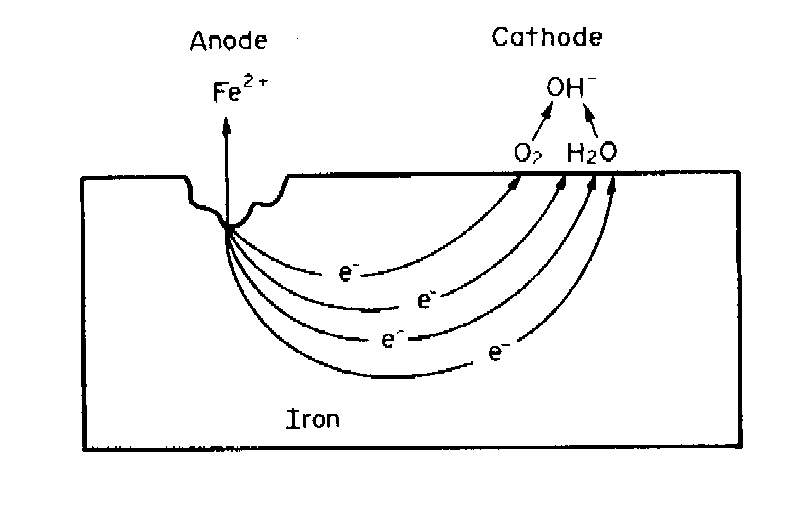
The sites where the anodic and cathodic reactions take place, the anodes and the cathodes of the corrosion cell, are determined by many factors: (i) they are not necessarily fixed in location; (ii) they can be adjacent or widely separated so that, for example, if two metals are in contact, one metal can be the anode and the other the cathode, leading to galvanic corrosion of the more anodic metal; (iii) there can exist variations over the surface of oxygen concentration in the environment that result in the establishment of an anode at those sites exposed to the environment containing the lower oxygen content – differential aeration corrosion; (iv) or similarly, variations in the concentration of metal ions or other species in the environment, arising because of the spatial orientation of the corroding metal and gravity; or finally, (v) variations in the homogeneity of the metal surface, due to the presence of inclusions, different phases, grain boundaries, disturbed metal, and other causes, can lead to the establishment of anodic and cathodic sites.
The process occurring at the anodic sites is the dissolution of metal as metallic ions in the electrolyte or the conversion of these ions to insoluble corrosion products such as rust. This is the destructive process called corrosion. The flow of electrons between the corroding anodes and the non-corroding cathodes forms the corrosion current, the value of which is determined by the rate of production of electrons by the anodic reaction and their consumption by the cathodic reaction. The rates of electron production and consumption, of course, must be equal or a buildup of charge would occur.
Driving force, corrosion tendenciesA driving force is necessary for electrons to flow between the anodes and the cathodes. This driving force is the difference in potential between the anodic and cathodic sites. This difference exists because each oxidation or reduction reaction has associated with it a potential determined by the tendency for the reaction to take place spontaneously. The potential is a measure of this tendency.
Since most corrosion reactions occurring in the presence of a liquid, such as water, are not chemical but are electrochemical, the use of chemical equilibria is of minimal use for studying aqueous corrosion. A useful way to study the relation of potential to corrosion is through the use of an electrochemical equilibrium diagram – called the "Pourbaix Diagram" – developed by M. Pourbaix, ( Available on the WWW) it plots potential against pH, a parameter also of great importance to corrosion processes.
|

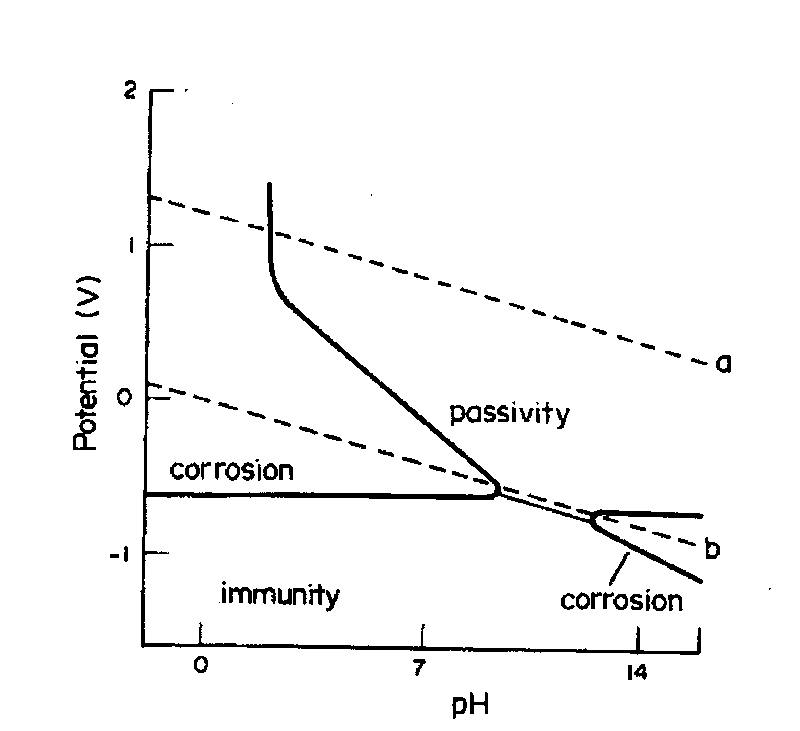 |
| Fig. 2. The potential – pH (Pourbaix ) equilibrium diagram for iron. |
Kinetics, corrosion rates
While it is necessary to determine corrosion tendencies by measuring potentials, it will not be sufficient to ascertain whether a given metal or alloy will suffer corrosion under a given set of environmental conditions. Because even though the tendency for corrosion may be high, the rate of corrosion may be very low, and thus corrosion may not be a problem.Corrosion rates are determined by applying a current to produce a polarization curve (the degree of potential change as a function of the amount of current applied) for the metal surface whose corrosion rate is being determined. When the potential of the metal surface is polarized by the application of the current in a positive direction, it is said to be anodically polarized; a negative direction signifies that it is cathodically polarized. The degree of polarization is a measure of how the rates of the anodic and the cathodic reactions are retarded by various environmental (concentration of metal ions, dissolved oxygen, etc. in solution) and/or surface process (adsorption, film formation, ease of release of electrons, etc.) factors. The former is called concentration polarization the latter activation polarization. The variation of potential as a function of current (a polarization curve) allows one to study the effect of concentration and activation processes on the rate at which anodic or cathodic reactions can give up or accept electrons. Hence, polarization measurements can thereby determine the rate of the reactions that are involved in the corrosion process – the corrosion rate.
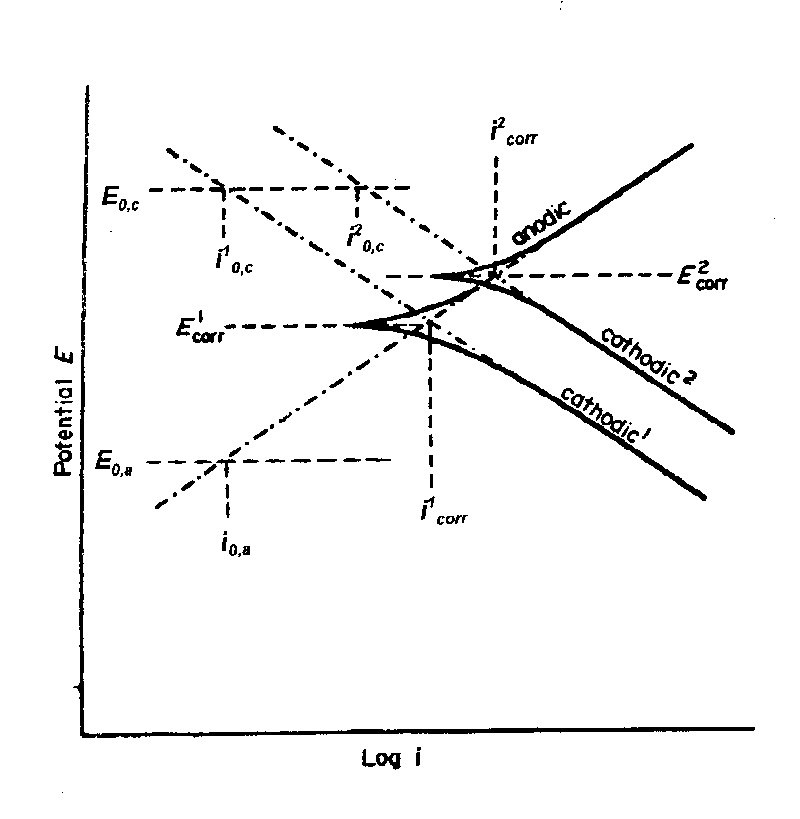 |
| Fig. 3 Schematic illustration of plots of potential "E" against the logarithm of the current density "i" (polarization curves) produced by making the investigated metal surface an anode and then a cathode. Two cathodic reactions are shown, the one with the higher cathodic exchange density "i2o,c" giving the higher corrosion current "i2corr". The extrapolations of the anodic and cathodic curves produce the Evans diagrams (see Figure 4). |
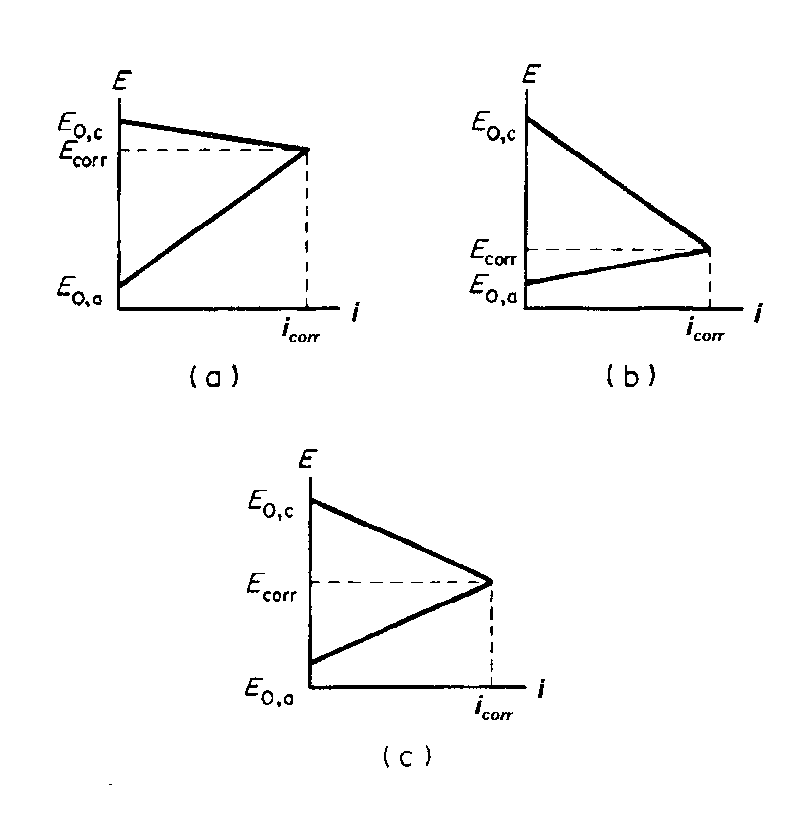 |
| Fig. 4. Evans diagrams (produced from the extrapolation shown in Figure 3, illustrating three main ways in which the corrosion rate can be mainly controlled: (a) control by the reactions on the anode; (b) control by the reactions on the cathode; and (c) control by a mixture of both. |
Parts of the extrapolations shown in Figure 3 produce the diagrams (Evans diagrams) shown in Figure 4. The relative values of the "slopes" of the anodic or cathodic polarization curves, determine whether the anodic, cathodic, or both reactions control the rate of the corrosion process. Evans diagrams are used extensively by corrosion scientists and engineers to evaluate the effect of various factors on corrosion rates.
 Passivity and the breakdown of passivity
Passivity and the breakdown of passivity
Since both driving force (corrosion-tendency) and kinetic (corrosion-rate) considerations are crucial in determining the extent of corrosion of a metal surface, a crucial factor controlling the rate is the existence of the phenomenon of passivity for certain metals and alloys, such as stainless steels and titanium. Considerable controversy still exists about what process is responsible for the existence of the phenomenon of passivity, even though many studies were carried out to develop a better understanding of it. For the purposes of this discussion, it is sufficient to characterize passivity as the conditions existing on a metal surface, because of the presence of a protective film whose nature is the source of the controversy, that markedly lower the rate of corrosion, even though active corrosion would be expected from driving force (corrosion-tendency) considerations.
To be effective, a passive film must provide a protective barrier that keeps the corrosion current on a metallic surface at a low enough value so that the extent of corrosion damage is minimized. An effective film is one that resists the breaching (called the breakdown) of the passive film. Breakdown processes lead to the forms of localized corrosion that are some of the major sources of corrosion failures, pitting, crevice, intergranular, and stress corrosion (see below). They cause the disruption of the passive film and thus expose discrete bare sites on the metal surface to an environment where the tendency for attack is very high. The many mechanisms proposed for chemical breakdown are based on two models, each having many variations. They are (i) Adsorbed Ion Displacement Models and (ii) Ion Migration or Penetration Models. Two types of breakdown processes exist: electrochemical breakdown where many studies have found that breakdown takes place at a potential above a specific "breakdown potential," and mechanical breakdown which occurs when the passive film is ruptured as a result of stress or abrasive wear. It should be noted that all breakdown mechanisms involve a damaging species. Unfortunately, one of the major species causing breakdown of passivity is the chloride ion, a damaging species abundantly available in nature.
Competing with the passive film breakdown process is the passive film repassivation process. Thus, an effective alloy for resisting localized corrosion would be one whose surface not only forms a passive film that resists the process leading to breakdown but also is capable of repassivating at a rate sufficiently high so that, once breakdown has occurred, exposure to a corrosive environment is minimal.
Types of corrosion
The breakdown and repair of passivity process just described is involved in many but not all of the various types of corrosion.
Pitting corrosion
The initiation of a pit occurs when electrochemical or chemical breakdown exposes a small local site on a metal surface to damaging species such as chloride ions. The sites where pits initiate are not completely understood, but some possibilities are at scratches, surface compositional heterogeneities (inclusions), or places where environmental variations exist. The pit grows if the high current density – the area of breakdown initiation is exceedingly small – involved in the repassivation process does not prevent the formation of a large local concentration of metal ions produced by dissolution at the point of initiation. If the rate of repassivation is not sufficient to choke off the pit growth, two new conditions develop. First, the metal ions produced by the breakdown process are precipitated as solid corrosion products (such as the Fe(OH)2, Appendix, Equation [7]) which usually cover the mouth of the pit. This covering traps the solution in the pit and allows the buildup of positive hydrogen ions through a hydrolysis reaction (Appendix, Equation [5]). Then, chloride or another damaging negative ions diffuse into the pit to maintain charge neutrality. Consequently, the repassivation becomes considerably difficult because the solution in the pit is highly acidic, contains a large concentration of damaging ions and metallic ions, and contains a low oxygen concentration. Thereby the rate of pit growth accelerates.
The pit is the anode of an electrochemical corrosion cell, and the cathode of the cell is the non-pitted surface. Since the surface area of the pit is a very small fraction of the cathodic surface area, all of the anodic corrosion current flows to the extremely small surface area of the breakdown initiation site. Thus, the anodic current density becomes very high, and penetration of a metal structure bearing only a few pits can be rapid.
Crevice corrosion
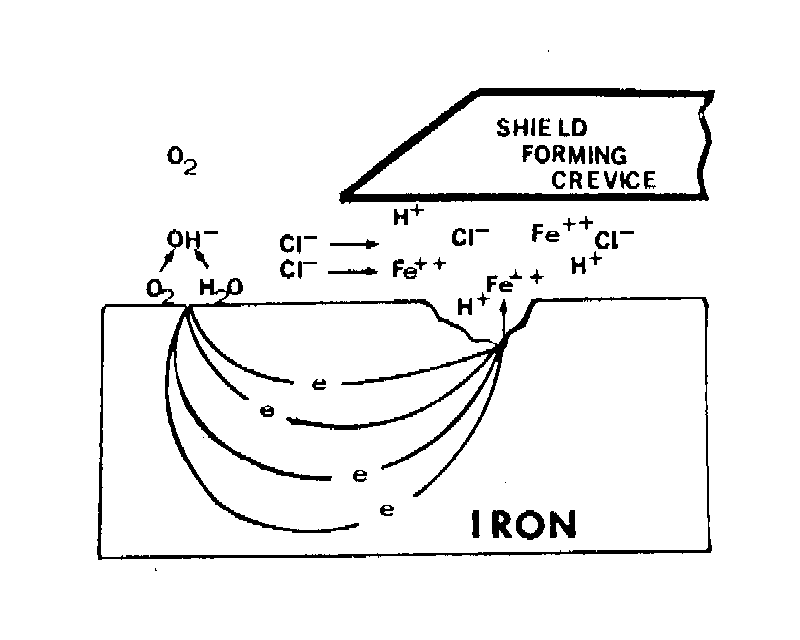 |
| Fig. 5. An idealized picture of the environment that develops in a crevice by the corrosion cell produced on iron by an anode in a crevice and a cathode outside of the crevice. |
Stress corrosion cracking, hydrogen damage, and corrosion fatigue
Stress corrosion cracking (SCC) is a form of localized corrosion which produces cracks in metals by the simultaneous action of a corrodent and tensile stress. The electrochemical cell between the exterior and the interior environments of a crack is similar to that described above for a crevice. Because of the necessity for the application of stress, the breakdown of the passive layer on a metal surface in SCC is generally ascribed to mechanical causes but many suggest that electrochemistry is a significant factor because it controls the rate of repair of the passive layer ruptured by mechanical stress.
Corrosion fatigue has a somewhat similar mode of failure to SCC. It mainly differs from SCC in that it involves situations where the stress is applied cyclically rather than as a static sustained tensile stress. With both corrosion fatigue and SCC, the issues of film mechanical breakdown, repassivation, adsorption of damaging species, hydrogen embrittlement, and electrochemical dissolution are to greater or lesser extents involved in determining susceptibility.
Intergranular corrosion
With the exception of metallic glasses, the metals used in practical devices are made up of small crystals (grains) whose surfaces join the surfaces of other grains to form grain boundaries. Such boundaries or the small regions adjacent to these boundaries can under certain conditions be considerably more reactive (by being more anodic) than the interior of the grains. The resulting corrosion is called intergranular corrosion. It can result in a loss of strength of metal part or the production of debris (grains that have fallen out).
Uniform corrosion
Uniform corrosion results from the sites, not necessarily fixed in location, that are distributed over a metal surface where the anodic and cathodic reactions take place. Uniform corrosion damage, sometimes called wastage, is usually manifested in the progressive thinning of a metal part until it virtually dissolves away or becomes a delicate lace-like structure.
Galvanic (and thermogalvanic) corrosion
Two metals having different potentials in a conducting electrolyte result in the more anodic metal usually being attacked by galvanic corrosion. Because of electrical contact between two different metals, galvanic corrosion differs from the other forms of corrosion described previously in that the anodic and cathodic sites of the corrosion cell reside separately on the two coupled different metals comprising the corrosion cell, while for the other forms of corrosion, the cathodes and anodes exist on the surface of the same metal.
The value for the differences in potential between two dissimilar metals is usually obtained from a listing of the standard equilibrium potentials for the various metals (for reactions of the type given in Appendix, Equation [1]). However, since the standard potentials assume bare metal surfaces in a standard solution containing their ions at unit concentration (strictly speaking, activity), while in practice metals invariably have films on their surfaces and are exposed to nonstandard environments, these potentials are not always reliable guides to the corrosion tendencies of the anodic member of a corrosion cell produced by two coupled dissimilar metals. Another factor besides the coupling of two different metals that can lead to galvanic corrosion is a difference in temperature at separated sites on the same metal surface. Such a situation leads to thermogalvanic corrosion. This kind of corrosion can be encountered in heat exchanger systems where temperature differences are common.
Selective leaching
When one component of an alloy is removed selectively by corrosion because there exist differences in potential between different constituents of an alloy – a situation analogous to that of the galvanic corrosion of coupled dissimilar metals. This results in an increase in the concentration of the remaining components and selective leaching occurs. This type of corrosion is also called parting and, after the most common example, the selective leaching out of zinc from brass, dezincification.
Erosion corrosion
Erosion corrosion result from the disruption of protective passive films by erosive or abrasive processes. Once the protective or passive film is removed in an aqueous electrolyte, the electrochemical processes for the types of corrosion described above take place.
Corrosion prevention
The types of electrochemical corrosion just described can be prevented or controlled by utilizing the current understanding of the principles underlying corrosion processes. This understanding has been the basis for the development of a number of corrosion prevention measures.
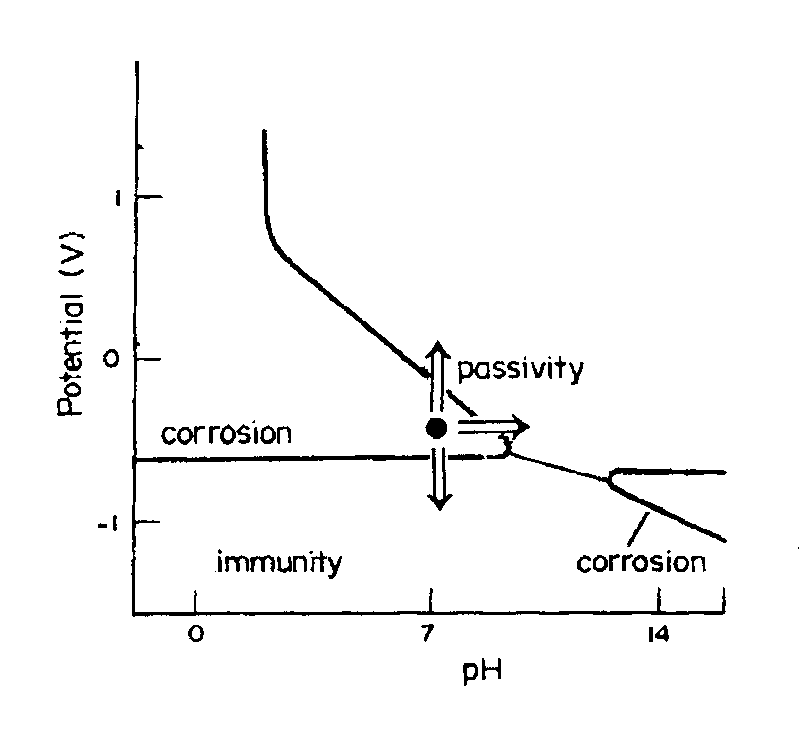 |
| Fig. 6. Pourbaix diagram for iron showing how corrosion protection can be achieved by using measures that bring a corroding system into either the immunity or the passivity region. |
Other measures used to control electrochemical corrosion processes are the following: (i) Corrosion resistant alloys are developed to be corrosion resistant and contain constituents such as chromium, which produce more effective protective films that resist breakdown and repassivate rapidly. (ii) Coatings are an artificial barrier to the corrosive environment on a metal surface as contrasted to the naturally formed barrier that a corrosion resistant alloy provides. The most widely used organic coatings are paints. Other organic coatings include lacquers, waxes, and greases. Some metallic coatings such as chrome plate provide a protective barrier because they are more corrosion resistant than the metals they protect. Zinc coatings, however, act in the opposite manner. This coating, because it has a greater tendency to corrode than the metal to which it is applied, cathodically protects it. Finally, ceramic coatings such as porcelain enamels and glasses are used to protect metal surfaces by providing chemically resistant barriers, usually oxides, which are more stable than metals. (iii) Corrosion inhibitors are added to the environment in which a given metal must be used to make the environment less corrosive. The inhibitors act either by adsorbing on metal surfaces by providing barriers to the environment or by keeping the environment from becoming more corrosive by providing a buffering action.
Corrosion rections
Some examples of the anodic and cathodic reactions that occur simultaneously on a metal surface in a "corrosion cell" are as follows.
A typical anodic oxidation that produces dissolved ionic product, for example for iron metal is:
[1] ![]() Fe ==> Fe2+ + 2e-
Fe ==> Fe2+ + 2e-
Examples of cathodic reductions involved in corrosion process are:
[2] ![]() O2 + 2H2O + 4e- ==> 4OH-
O2 + 2H2O + 4e- ==> 4OH-
[3] ![]() O2 + 4H+ + 4e- ==> 2H2O
O2 + 4H+ + 4e- ==> 2H2O
[4] ![]() 2H+ + 2e- ==> H2
2H+ + 2e- ==> H2
The cathodic reaction represented by Equation [2] exemplifies corrosion in natural environments where corrosion occurs at nearly neutral pH values. Equations [3] and [4] represent corrosion processes taking place in the acidic environments encountered in industrial processes or for the confined volumes (pits, crevices) where the pH can reach acidic values because of hydrolysis reactions such as:
[5] ![]() Fe2+ + 2H2O ==> Fe(OH)2 + 2H+
Fe2+ + 2H2O ==> Fe(OH)2 + 2H+
This reaction produces H+ ions, the concentration of which can, uder certain conditions, become large if the H+ ions cannot readily move out from a confined volume. The overall corrosion reaction is, of course, the sum of the cathodic and anodic partial reactions. For example, for a reaction producing dissolved ions (sum of reactions [1] and [4]):
[6] ![]() Fe + 2H+ ==> Fe2+ + H2
Fe + 2H+ ==> Fe2+ + H2
or, for a reaction producing insoluble hydroxide (sum of reactions [1] and [2]):
[7] ![]() 2Fe + O2 + 2H2O ==> 2Fe(OH)2
2Fe + O2 + 2H2O ==> 2Fe(OH)2
The Tafel equation is an early (1905) empirical relation between the overpotential of the electrode and the current density passing through the electrode:![]() Tafel equation
Tafel equation
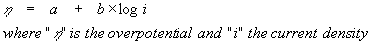
"a" and "b" are characteristic constants of the electrode system. A plot of electrode potential against the logarithm of the current density is called the "Tafel plot" and the resulting straight line the "Tafel line". "b" is the "Tafel slope" that provides information about the mechanism of the reaction, and "a" the intercept, provides information about the rate constant (and the exchange current density) of the reaction.
Related articles
AnodizingArchaeological use of electrochemistry
Cathodic/anodic corrosion protection
Corrosion inhibition
Bibliography
- Principles and Prevention of Corrosion (2nd edition), D. A. Jones, Prentice Hall, Upper Saddle River NJ, 1996.
- Corrosion (two volumes), L. L. Shreir, R. A. Jarman, and G. T. Burstein (editors), Butterworth-Heinemann, Oxford, Boston, 1994.
- Advances in Localized Corrosion: NACE-9, H. S. Isaacs, U. Bertocci, J. Kruger, and S. Smialowska (editors), National Association of Corrosion Engineers, Houston TX, 1990.
- Passivity of Metals – a Materials Science Perspective, J. Kruger,
"International Materials Reviews" Vol. 33, pp 113-129, 1988.
- Corrosion Engineering (2nd edition), M. G. Fontana, McGraw-Hill, New York, 1986.
- Corrosion and Corrosion Control: An Introduction to Corrosion Science and Engineering (3rd edition), H. H. Uhlig and R. W. Revie, Wiley, New York, 1985.
- Basic Corrosion and Oxidation, J. M. West, Ellis Horwood, Chichester UK, 1980.
- Passivity of Metals, R. P. Frankenthal and J. Kruger (editors), The Electrochemical Society, Pennington NJ, 1978.
- Atlas of Electrochemical Equilibria in Aqueous Solutions (2nd edition), M. Pourbaix, National Association of Corrosion Engineers, Houston TX, 1974.
Listings of electrochemistry books, review chapters, proceedings volumes, and full text of some historical publications are also available in the Electrochemistry Science and Technology Information Resource (ESTIR). (http://knowledge.electrochem.org/estir/)
Return to: Top – Encyclopedia Home Page – Table of Contents – Author Index – Subject Index – Search – Dictionary – ESTIR Home Page – ECS Home Page
ECS | Redcat Blog | ECS Digital Library