Return to:
Encyclopedia Home Page –
Table of Contents –
Author Index –
Subject Index –
Search –
Dictionary –
ESTIR Home Page –
ECS Home Page
THE PAST, PRESENT, AND FUTURE OF ELECTROANALYTICAL CHEMISTRY
Peter T. Kissinger
Purdue University and Bioanalytical Systems, Inc.
West Lafayette, Indiana, USA
(May, 2005)
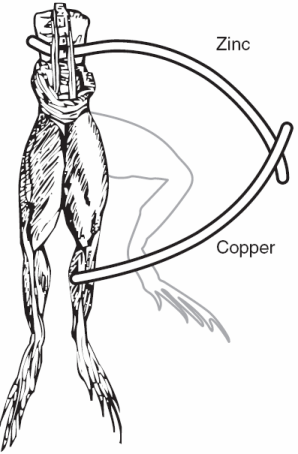 |
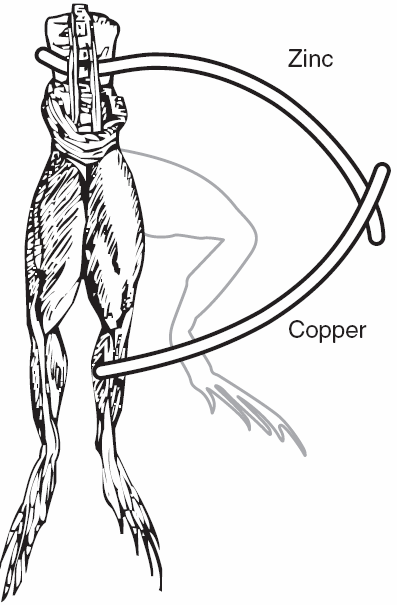
| Fig. 1. Galvanic cell circa 1786, electrochemistry and electrophysiology began with frogs' legs. |
Luigi Galvani got electrochemistry started in the mid 1780s. A lot could be accomplished with frogs' legs and two different metals (Figure 1). After the day's experiments, a gourmet meal was readily prepared. Galvani's work is generally considered to be the beginning of electrophysiology. It is now widely appreciated that biology has a strong electrochemical component. Cell membranes are "charged" and the nervous system in all of us consists of circuits carrying electrical activity along cells called neurons (the "wires" and "connectors" from the simple worm or snail all the way to a human). You can see the resemblance between nerve cells and complex electrical wiring in Figure 2.
 |
| Fig. 2. Neurons are electrochemical networks. |
In the late 1790s, Alessandro Volta took away the frogs, and used other sources of salt water. His "pile" made electrochemistry really take off. He introduced what we now consider to be a battery. The article on Volta in this Encyclopedia clearly shows why "pile" was used to describe this innovation. Jargon is nothing new to science. The term "battery" was introduced by Benjamin Franklin, who also defined the electrical context for plus and minus, positive or negative, and conductor. Nicholson and Carlisle used Volta's pile (1800) to demonstrate the electrolytic decomposition of water into hydrogen and oxygen gas. Humphrey Davy then (circa 1807) used it to carry out electrochemistry on molten salts and thereby isolate the alkali metals and alkaline earths in their elemental forms. By 1834, Faraday had pretty well worked out what was going on, and the rest of us have been trying to fill in some of the details ever since.
The chemistry profession can be divided into categories based on making something happen and characterizing what happened. Examples of the former would include making a plastic, a lubricant, a drug, a paint, an alloy, an explosion. Examples of the latter include how much, what concentration, how fast, how hot, what pressure, what energy. These questions suggest numerical answers that we call data. Obtaining data about chemical things is the focus of analytical chemistry, just as obtaining data about companies is the job of a Wall Street analyst. Good data is said to be valid and auditable. Otherwise, it is bad data.
If we narrow chemistry down to the more specific (but still very broad) field of electrochemistry, we can look at examples of making something happen:
We can characterize what happened by answering questions such as these:
Answering questions like these is the province of electroanalytical chemistry. They imply the need for measurement tools or electrochemical instruments.
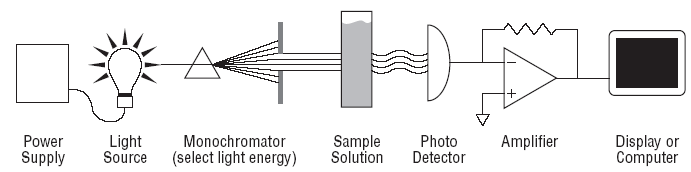
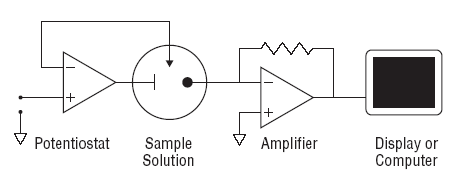
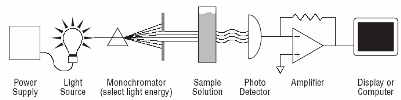
From an analytical chemistry perspective, the best feature of electrochemistry is the direct conversion of chemical information into an electrical signal. There is no need for magnets, light bulbs, lasers, or vacuum pumps. Keep in mind that all matter is electrical, all atoms in molecules have electrons. If we can grab these electrons, their energy can tell us what we are looking at and their number can tell us how much of that is present in a sample under test. Most analytical chemistry instruments use light or magnetism. Light is the easiest to understand for this application. By just looking at a glass of fruit juice, we can get a good idea of what fruit was used (yellow for apple juice, red for cranberry juice) and how much is present (a darker color indicates a greater concentration). We use the eye, but all of this can be done more precisely and automatically using instruments called spectrophotometers coupled to a small computer. A precisely controlled light bulb replaces sunlight and a solid state detector replaces the human eye. Take a look at Figure 3. For analytical electrochemistry, much of this can be simplified as in Figure 4, where there is no light source, and power requirements can be as modest as a cellular telephone battery. Because electrochemistry depends on a surface phenomenon, not an optical path length, the sample volume can be very small.
 |
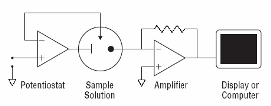 |
| Fig. 3. Simple analytical spectroscopy involves monitoring light intensity after it passes through a sample of three dimensions. |
Fig. 4. Simple amperometric electrochemical devices monitor current produced in response to an electrical potential applied between an electrode surface and a solution. No bulky light sources or monochromators are required. Electrochemistry is a surface methodology, making it easy to study events in very small volumes. |
As we transitioned into the 20th century, there was virtually no concept of "an electroanalytical technique". Some basic ideas were established by Sand and Cottrell, but it was not until Heyrovsky's polarography (the 1959
Chemistry Nobel Prize winner) work in the 1920s that electrochemical analysis could really be taken seriously. On the other hand, the concept of electrochemistry as a means of power generation was very well established by that time. Edison cells were widely used in commerce. It is fair to say that electroanalysis did not really have that much of an impact until after World War II. After the war, the scope of electronics rapidly moved beyond radio and even into chemistry labs. This enabled Faraday's Law, the Nernst equation, and Heyrovsky's "polarograms" to advance beyond curiosities into widespread consideration. We thus might come to the (somewhat imprecise) conclusion that we have had roughly half a century of electroanalytical chemistry as a popular workhorse of analytical chemistry. This includes ion selective electrodes (the pH meter to start), the now classical "electrometric" titrations (potentiometric, amperometric, and coulometric), the notion of a "biosensor," and the concept of voltammetry as a broadly based tool for obtaining data about energetics, kinetics, concentration, and reaction mechanism.
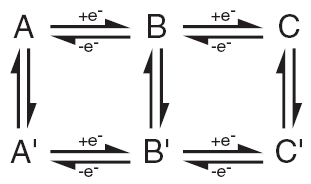 |
| Fig. 5. Electrochemists spend a lot of time figuring out mechanisms of chemical reactions coupled to electron transfer steps. The rates and energetics of each step are sought. |
I realize that many of the words in the last several lines will mean nothing to most readers. Jargon is everywhere. I do want to give a little attention to "reaction mechanism," which is also referred to as a "reaction pathway" or even a "channel". These all imply there is often more than one way to get from a reactant ("starting material") to a product using electrochemistry. To study a reaction mechanism is to understand which steps are taken or path is followed to arrive at the final product. In Figure 5, you can see a "scheme of squares" that relates different chemical species by the gain or loss of electrons along alternate pathways. Electrochemical steps are shown horizontally and purely chemical steps (not involving electron exchange) are shown vertically.
Prior to 1960, electrochemical instrumentation consisted mainly of Wheatstone bridges, light beam galvanometers, motor-driven wire-wound potentiometers, vacuum tube electrometers, and analog panel meters. Even in the 1950s (when radar, rockets, and televisions were well advanced), electrochemists were using burettes and balances not different in concept from those of Faraday. While potentiostats were known to exist before 1960 and the first vacuum tube operational amplifier circuits (analog computers) were applied to electrochemistry in the late 1950s, it was not until the late 1960s that three-electrode instrumentation became common on a global scale. A potentiostat has the purpose of maintaining the potential of an electrode constant ("stat") as the current across the electrode-solution junction varies. This made electrochemical studies in poorly conducting solutions very feasible, greatly expanding the range of analytical electrochemistry in the 1960s.
At that crucial point, progress was accelerated with solid-state operation amplifiers in common use by 1965 and (somewhat fragile) integrated operational amplifiers by 1970. Solid-state pH meters were rare before 1970 and the battery powered CMOS LCD models (available today for under $200.00 US) certainly did not exist at this time. Solid-state pH meters are now available in aquarium stores and some garden shops. These are not laboratory quality instruments, but they are adequate to the task of managing fish or growing vegetables. Computer-based instrumentation followed a rapid progression from Digital Equipment PDP-8s (1960s) to Apple IIs (1970s), to the still evolving "IBM PC�. What is next?
Electrochemical instrumentation, over the last 50 years, has tagged along behind developments in electronics and software intended for military and consumer applications. Economics continues to dictate this because within the already small instrumentation market, electrochemistry is microscopic. The only big time product remains the pH meter. To predict the future, one must look to telecommunications, including the integration of video with computers, worldwide wireless communications, advanced software development tools, and further miniaturization of hardware by great integration of digital and analogue modules. Even now we can use processor-based instrumentation to carry out an array of electrochemical techniques on a given sample. One of the most productive aspects of this approach is the fact that multiple users can employ an electrochemical workstation without interfering with each other. In the past, instruments were rebuilt or re-adjusted for different electrochemical methods. This adjustment is now totally in the hands of software.
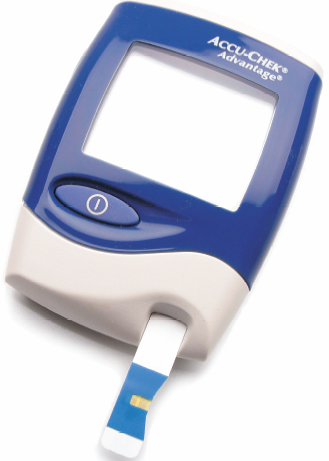 |
| Fig. 6. An amperometric blood glucose instrument of the type available at pharmacies worldwide (Roche Diagnostics). Only a few microliters (millionth of a liter) of blood are required. |
I do not predict a revolution in the next decade, but a steady evolution of what we already have been doing. Software will become easier to develop and easier to use, but the low volume will continue to require premium pricing to pay developers (Windows operating systems sell ten million units per year, digital electrochemical instruments sell hundred units per year). Automation of sample processing will finally become practical for voltammetric analysis. Throwaway electrodes are already in use for glucose sensing and will clearly find other uses in the years ahead. A neat way to see quite advanced electroanalytical instruments is to go to any pharmacy and look for glucose meters in the section on diabetes care. You will see a device in a small plastic case with a digital display. These meters are today roughly the size of the smallest cellular telephone. They include data storage for many measurements over time. Trend information is important to diabetics and their physicians. Along with the meter, you buy disposable electrochemical cells containing 2-3 electrodes on a plastic strip that plugs into the meter. A drop of blood, or even 1/25 of a drop of blood, is all that is required for these electrochemical cells to work. Everything is automatic once the sample is applied. If you have not seen one of these electroanalytical tools, go to Wal-Mart or any pharmacy and take a look. Figure 6 shows one of these meters with its "plug in" disposable electrode strips. There are many competitors battling for market share, making the technology better and better each year. The current market is about $2 billion/year! It is fair to say that this one application is responsible for over 95% of the total market for all amperometric instruments and supplies sold each year.
Rapid digital simulation of electrochemical mechanisms has had an enormous impact on teaching and understanding the chemistry behind (often surprising) complex current voltage curves. The work of Manfred Rudolph in Jena, Germany took a 30-year-old concept and made it practical for every chemist. His electrochemical simulation algorithm is already a leading video game among scientists. It will remarkably assist in the design of real experiments and the explication of previously incomprehensible voltammograms. The relationship of time, space, concentration, current, and potential are all made evident to the experimenter within a few seconds. Yes. Simulation is an artificial representation of a real event, a model of reality based on mathematical rules for basic steps. We have computers simulating battles, football games, space flight, chess games, and electrochemical processes.
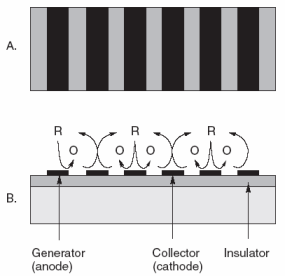 |
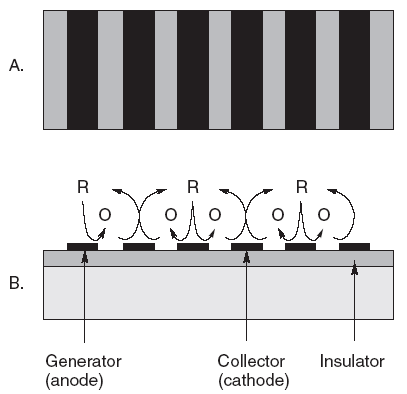
| Fig. 7. Interdigitated arrays of electrodes made with modern lithography techniques have been interesting materials for modern electrochemical studies of redox reactions (A) top view, (B) side view. The reduced form represented by R is oxidized to O at the anodes, while O is recycled to R at the alternating cathodes. There are many applications for such a scheme. |
For 25 years or so, the instrumentation has not been limiting. The "materials science" aspects of electrodes have been limiting. Access to more reproducible and reliable electrodes is the rate-controlling step to more widespread use of electrochemical analysis. The electronics are already well ahead. With all of the work in chemically modified electrodes and biosensors for over 30 years, we still do not have many surfaces we can trust to behave themselves. Surfaces are very delicate on molecular dimensions and can easily be altered in ways that are difficult to predict. While many academics have continued to identify interesting topics to explore, for example, interdigitated arrays, single crystals, and electrodes of 0.1 µm; these have not yet helped us solve many practical problems for the world at large. See Figure 7 for an example of an interdigitated (interlocking fingers) array of electrodes.
As science and technology have progressed since 1800, we inevitably make advances of increasingly narrow significance until individual investigators work on problems and use tools only of interest to themselves. In our instrument business, this becomes a problem for technology transfer in that only a small percentage of academic innovation finds a market of sufficient size. The popular press often accuses academic scientists of pursuing a hobby at the taxpayers' expense. This stings because there is some element of truth to it. An amazing number of articles in the expanding number of journals are read by few beyond the authors. It is difficult to justify this simply on the basis that something great could result when smart people do research. Nevertheless, history tells us that is what happens!
The process of carrying out truly significant advances is not easily controlled or predicted. Scientists and engineers today must work hard to be well read and broadminded in their thinking. Those who know what they do not need to know and learn what they do not need to learn will be the most successful; others will be hopelessly narrow. Humphrey Davy, Michael Faraday, and Walther
Nernst (the 1920
Chemistry Nobel Prize winner) should be an inspiration to us. Natural philosophy was their calling and electrochemistry was just a small part of it. Practical and fundamental problems were considered to be one and the same. It is often developments from cross fertilization between diverse fields that are truly significant. Thus, it is foolish to consider electrochemistry as a subject that is independent of biochemistry, solid-state physics, optics, or mathematics. For example, the autofocus 35 mm camera is an electrochemical device, as is the notebook computer and cellular phone. Electrodes are used to relieve pain in dental surgery and to help bone fractures heal in medicine. Over 25 firms have manufactured electrochemical detectors for liquid chromatography. Many of these are used to study how the human brain works.
Galvani did not predict Volta's findings, Volta did not predict Faraday's work, Faraday did not predict Nernst's discoveries, but my prediction is that electroanalytical instrumentation is well in hand now and is waiting for the surface chemistry to come under greater control. Uniformly prepared and hermetically packaged, film electrodes might just give us that disposable electrode that we have always wanted ever since Heyrovsky (the 1959
Chemistry Nobel Prize winner) demonstrated what a clean electrode can do. Owing to the unpredictable non-linear behavior of electrodes under many circumstances, instrumentation and computer algorithms do not often compensate for a "bad" electrode. Is nanotechnology a possible solution? Time will tell.
The following Appendix provides a little more detail. It requires some understanding of chemistry and physics (electricity). It is fair to say that a working knowledge of electroanalytical chemistry absolutely requires some knowledge of voltage, current, and charge.
Appendix
ELECTROCHEMISTRY FOR THE NON-ELECTROCHEMIST
Peter T. Kissinger and Adrian W. Bott
Bioanalytical Systems, Inc.
West Lafayette, IN 47906, USA
This appendix discusses nine basic concepts in finite current electrochemistry.
 Electrodes are surfaces Electrodes are surfaces
The fundamental process in electrochemistry is the transfer of electrons between the electrode surface and molecules in the solution adjacent to the electrode surface; that is, it is a heterogeneous process, in contrast to many other measurements (for example, NMR, UV, etc) that typically involve homogeneous solutions in three dimensions. Therefore, it is often useful to consider quantities per unit area as well as per total area. One quantity that is particularly important in electrochemical experiments is current "i" and the current/per unit area is referred to as the current density (amp/cm2). The current density is the more fundamental quantity, since it is independent of the electrode surface area, whereas the current is proportional to the electrode surface area.
Since the current results from a chemical substance dissolved in the solution adjacent to the electrode surface, it is not surprising that the probability of a molecule or ion reacting at the electrode surface is directly related to the concentration. Therefore, the current density is directly proportional to the concentration, provided the electrode reaction does not have unusual features, such as adsorption and dimerization).
(A point of nomenclature: It is redundant to say that "current flows". The word current is sufficient. When water or charge flows, we have current.)
Current is rate
Current is expressed in amperes; that is, coulombs/second. As the word implies, it is a quantitative expression of how fast something is happening. The differential form of Faraday's Law
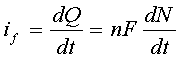
shows that the rate at which electrons are moved across the electrode-solution interface (that is, the faradaic current "if") is directly related to the rate of the reaction that is occurring at that interface. (Here, "Q" is the charge, "t" is time, "n" is the number of electrons transferred per molecule, "F" is the Faraday number, and "N" is the number of moles of material processed by the electrochemical reaction.) Therefore, when we measure the faradaic current, we are also measuring the rate of a chemical reaction. This is a unique feature of electrochemistry. Most other techniques do not provide an instantaneous measure of rate.
It should be noted that there are multiple steps involved in an electrochemical reaction, such as the electron transfer reaction, the transport of molecules from the bulk solution to the electrode surface, and chemical reactions coupled to the electron transfer reaction. As with any multi-step reaction, the rate of the overall reaction is generally determined by the rate of the slowest step (the rate determining step), and it is important to know which step is the rate determining step. In analytical electrochemistry of dissolved species, this step is typically the transport of molecules to the electrode surface. However, there are many instances where this is not the case and where the rate of the heterogeneous electron transfer reaction is of interest (for example, corrosion electrochemistry).
As discussed, the electrode surface area and reactant concentration affect the current. Other factors that affect the current include the temperature and the viscosity of the solution, and it is important to ensure that these parameters remain constant, so that the current is proportional to the reactant concentration (this is particularly important for quantitative analysis).
There are some other sources of current in addition to the faradaic current due to the analyte, which are collectively referred to as the background current. These sources include the electrolysis of impurities, the electrolysis of the electrolyte, the electrolysis of the electrode material, and capacitive (or charging) current The first three of these are faradaic currents, whereas the last one is a property of the interface between the electrode and the solution. This interface behaves like an electrical capacitor in that it can store charge, and (to a first approximation) it obeys the equation:
Q = CV
where "Q' is the charge stored, "C" is the capacitance (in farads), and "V" is the potential difference across the interface. An equation for the capacitive current "ic" can be obtained by differentiating the above equation:
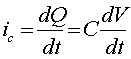
The total current will therefore be a summation of currents, including the faradaic currents for the analyte, the electrolyte, and the electrode material, as well as the capacitive current. All but the first of these currents are generally undesirable, and attention must be given to minimizing their effects. In some cases, the background current can be subtracted from the total current to leave the signal of interest. However, in other cases, the elimination of the background current is not so easy. In the worst cases, the background and signal currents are combined in a non-linear manner, making it impossible to reliably estimate the background current in the absence of the sample. Fortunately, such cases are rare. The capacitive current can be eliminated by operating the electrode at a fixed potential (that is, dV/dt = 0). One application where the background current can be essentially eliminated is liquid chromatography with electrochemical detection (LCEC), since a) the fixed potential eliminates the capacitive current and b) the background faradaic currents can be precisely measured between peaks, and then can be subtracted from the analyte signal. However, it is important to note that the capacitive current will change (even at a fixed potential) if the ionic strength or the solvent composition changes the bulk properties of the mobile phase. Such a change can be a transient disturbance; for example, at the "solvent front" or "void volume" on a liquid chromatogram.
Charge is amount
If we integrate a rate, we obtain an amount. From the integrated form of Faraday's Law,
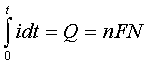
we can see that the total amount of electricity passed ("Q" the charge, measured in units of coulombs) is proportional to the number of moles "N" of material processed by the electrochemical reaction.
Potential controls the position of the equilibrium between O and R
The fundamental reaction in electrochemistry is the equilibrium between two species (O and R) that are interconverted by the transfer of an electron:
O + e- <==> R
When discussing the effect of the applied potential on this reaction (or redox couple), a useful analogy is the acid-base equilibrium
A- + H+ <==> HA
The position of the acid-base equilibrium is determined by the concentration of hydrogen ions, or protons, (that is, the pH), which can be considered in terms of a proton pressure (for example, increasing the proton pressure (decreasing the pH) moves the equilibrium to the right). When considering the electron transfer equilibrium, we can consider an analogous electron pressure, which determines the position of the equilibrium between and O and R. The applied potential is a measure of the electron pressure in the same way that pH is a measure of proton pressure.
In both cases, there is a unique situation in which the concentration of each form (O and R or A- and HA) are equal. This occurs at the potential/electron pressure "Eo'" in the former case and the pH/proton pressure pKa. These values provide a convenient index of the relative strength of an oxidant/reductant or acid/base. In both cases, the values are medium dependent (that is, they will differ between various non-aqueous and aqueous solutions).
When the pH is higher than the pKa, an acid will favor its protonated form. When the applied potential is more negative than "Eo'", a redox couple will favor the reduced form R.
Potential controls rate
There are two important parameters that affect the rate of the heterogeneous electron transfer reaction – the intrinsic ability of the molecule to undergo the electron transfer reaction (as reflected in the rate constant "ks") and the applied potential. The rate of electron transfer increases exponentially with the applied potential. Therefore, we can adjust the rate by varying the applied potential; for example, if the intrinsic rate of electron transfer for a given species is slow, it can be accelerated by applying a sufficiently large potential. Relatively more positive potential will speed up oxidations, whereas relatively more negative potentials will speed up reductions (note that, for example, a "relatively more negative potential" can still be positive; see section below).
The voltammetric axes
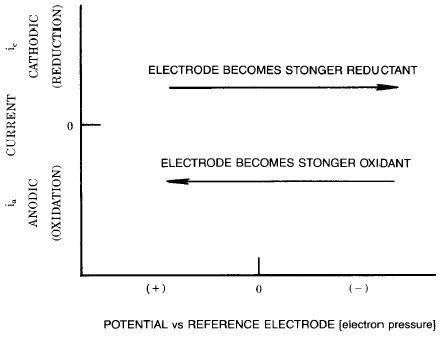 |
| Fig. a1. The classical (or polarographic) convention for plotting voltammetry data. |
The most common format for the presentation of electrochemical data is a plot of current "i" against potential "E". However, it is important to note that there are a number of different axis conventions. In the classical (or polarographic) convention, negative potentials are plotted in the positive "x" direction, and cathodic currents (due to reductions) are positive. In the IUPAC convention, positive potentials are plotted in the positive "x" direction, and anodic currents (due to oxidations) are positive (in other conventions, current is plotted along the axis and/or a logarithmic current scale is used). It is also vital to realize that the potential values are quoted relative to a reference potential (the value of which is defined by the reference electrode); that is, the zero on the x-axis is quite arbitrary. Therefore, there is no significance in the absolute values. Different values of "Eo'" for a given species under identical conditions are measured for different reference electrodes. It should also be noted that there is no correlation between positive and negative potentials and oxidations and reductions; that is, reductions can occur at positive potentials and oxidations can occur at negative potentials.
Current, potential, and time
There are only two basic formats for displaying experimental data; current "i" against potential "E" (voltammetry) and current against time "t" (chronoamperometry). (Charge plots are obtained by integrating current plots.) Since potential is often related to time by experimental design, "i" – "E" experiments also contain some features of the "i" – "t" experiments.
For historical reasons, more electroanalytical techniques have been developed than are really needed, and the large number of techniques can intimidate the novice. However, it should be noted that the vast majority of these are simple combinations of voltammetry and chronoamperometry. The key experimental variables in a given experiment are "E", "i", and "t". Only one of "E" and "i" can be varied independently, and "t" takes care of itself. The inverse relationship between time and frequency often plays a role (for example, high frequency experiments can be used to look at fast reactions), although the data from frequency domain experiments can be more difficult to interpret.
Light
The electrical measures described above are not all inclusive to electrochemical experiments. For example, electrode reactions can result in the emission of light (electrochemiluminescence). Likewise, light can also influence the rates and mechanisms of electrochemical reactions (photoelectrochemistry). Phenomena of this sort have been known since the 19th century. Finally light is often used to study electrochemical reaction mechanisms. This falls under the general terminology of spectroelectrochemistry. It ranges from the UV to the IR and includes transmission as well as reflection (off the electrode) methods. Raman spectroscopy of material adsorbed on electrodes is used as well as a variety of high vacuum techniques. The latter require removal of the electrode from solution (and are well beyond the scope of this primer).
The sound of one hand clapping
While it is obvious once given some thought, we should not forget that electrochemistry with a single electrode is like the sound of one hand clapping. In other words, it is largely meaningless. One needs a minimum of two electrodes to light a light bulb or to do electrochemistry. If a reduction (oxidation) is occurring at the working electrode, then a second electrode must be present and an oxidation (reduction) must be occurring there. The two electrodes are needed to "complete the circuit" (using the language of high school physics). Yes. You can have more than two electrodes. A reference electrode is often the third electrode, but it is also possible to have any number of working electrodes, all operating at different applied potentials in an array. Needless to say, this is not common.
Conclusion
It has been our intent in this appendix to outline nine of the most basic concepts of electrochemistry, since we have found that these concepts must be thoroughly mastered before more advanced topics can be understood. In our experience, students often become confused about various issues because these very fundamental ideas are not second nature to them.
Acknowledgement
The material of the Appendix was reproduced by permission from Current Separations, Vol. 20, No.2 (2002).
Related articles
Electrochemical blood glucose test
Electrochemical nose
Electrochemical sensors
Jaroslav Heyrovsky and polarography
Bibliography
- Broadening Electrochemical Horizons: Principles and Illustration of Voltammetric and Related Techniques, A. Bond, Oxford University Press, Oxford, 2002.
- Electrochemical Methods: Fundamentals and Applications (2nd edition), A. J. Bard and L. R. Faulkner, Wiley, New York, 2001.
- Organic Electrochemistry (4th edition), H. Lund and O. Hammerich (editors), Marcel Dekker, New York, 2001.
- Fundamentals of Electroanalytical Chemistry, P. M. S. Monk, Wiley, New York, 2001.
- Modern Electrochemistry 2B: Electrodics in Chemistry, Engineering, Biology and Environmental Science (2nd edition), J. O�M. Bockris and A. K. N. Reddy, Kluwer/Plenum, New York, 2000.
- Modern Electrochemistry 2A: Fundamentals of Electrodics (2nd edition), J. O�M. Bockris, A. K. N. Reddy, and M. E. Gamboa-Aldeco, Kluwer/Plenum, New York, 2000.
- Analytical Electrochemistry (2nd edition), J. Wang, Wiley, 2000.
- Modern Electrochemistry 1: Ionics (2nd edition), J. O�M. Bockris and A. K. N. Reddy, Kluwer/Plenum, New York, 1998.
- Electroanalysis (Oxford Chemistry Primers No. 64), C. M. A. Brett and A. M. O. Brett, Oxford University Press, Oxford, 1998.
- Electrochemistry, C. H. Hamann, A. Hamnett, and W. Vielstich, Wiley-VCH, Weinheim, Germany, 1998.
- Chemistry Is Electric!, M. E. Bowden, The Chemical Heritage Foundation, Philadelphia, 1997.
- Environmental Electrochemistry: Fundamentals and Applications in Pollution Abatement, K. Rajeshwar and J. G. Ibanez, Academic Press, New York, 1997.
- Electrode Potentials (Oxford Chemistry Primers No. 41), R. G. Compton, and G. H. W. Sanders, Oxford University Press, Oxford, 1996.
- Electrode Dynamics (Oxford Chemistry Primers No. 34), A. C. Fisher, Oxford University Press, Oxford, 1996.
- Laboratory Techniques in Electroanalytical Chemistry (2nd edition), P. T. Kissinger and W. R. Heineman (editors), Marcel Dekker, New York, 1996.
- Interfacial Electrochemistry, W. Schmickler, Oxford University Press, Oxford, 1996.
- Solid State Electrochemistry, P. G. Bruce (editor), Cambridge University Press, Cambridge, 1995.
- Physical Electrochemistry: Principles, Methods and Applications, I. Rubinstein (editor), Marcel Dekker, New York, 1995.
- Electrochemistry for Chemists (2nd edition), D. T. Sawyer, A. Sobkowiak, and J. L. Roberts, Wiley, New York, 1995.
- Techniques and Mechanisms in Electrochemistry, P. A. Christensen and A. Hamnett, Kluwer, Boston, 1994.
- Principles and Applications of Electrochemistry (4th edition), D. R. Crow, Blackie Academic and Professional, Glasgow, UK, 1994.
- Electrochemistry (2nd edition), P. H. Rieger, Kluwer, Boston, 1994.
- Electrochemistry: Principles, Methods, and Applications, C. M. A. Brett, and A. M. O. Brett, Oxford University Press, Oxford, 1993.
- Standard Potentials in Aqueous Solution, A. J. Bard, J. Jordan, and R. Parsons (editors), Marcel Dekker, New York, 1985.
- Theory and Application of Cyclic Voltammetry for Measurement of Electrode Reaction Kinetics, R. S. Nicholson, �Analytical Chemistry� Vol. 37, pp 1351-1355, 1965. Available on the WWW.
- Theory of Stationary Electrode Polarography: Single Scan and Cyclic Methods Applied to Reversible, Irreversible, and Kinetic Systems, R. S. Nicholson and I. Shain, �Analytical Chemistry� Vol. 36, pp 706-723 and 1212, 1964. Available on the WWW.
- �ber die Anwendung von Membranelektroden bei der Untersuchung von Ionenkonzentrationen, E. Pungor and E. Holl�s-Rokosinyi, �Acta Chimica Academiae Scientiarum Hungaricae� Vol. 27, pp 63-68, 1961. Available on the WWW.
- Studies in Electrode Polarisation. Part IV.-The Automatic Control of the Potential of a Working Electrode, A. Hickling, �Transactions of the Faraday Society� Vol. 38, pp 27-33, 1942. Available on the WWW.
- Apparatus for testing acidity, A. O. Beckman and . E. Fracker, �US Patent� No. 2,058,761, 1936. Available on the WWW.
- An improved, continuous-reading hydrogen-ion meter, K. H. Goode, �Journal of the American Chemical Society� Vol. 47, pp 2483�2488, 1925. Available on the WWW.
- Researches with the Dropping Mercury Cathode, Part I. General Introduction, Part II. The Polarograph, Part III. A Theory of Over-potential, J. Heyrovsky (and M. Shikata, Part II), �Recueil des Travaux Chimiques des Pays-Bas� Vol. 44 (Ser. 4, Vol. 6), pp 488-495, 496-498, and 499-502, 1925.
Available on the WWW.
- Electrolysis with a Dropping Mercury Catode. Part I. Deposition of Alkali and Alkaline Earth Metals, J. Heyrovsky, �The London, Edinburgh, and Dublin Philosophical Magazine and Journal of Science� Ser. 6, Vol. 45, pp 303-315, 1923.
Available on the WWW.
- Elektrolysa se rtufovou kapkovuo kathodou, J. Heyrovsky, �Chemicke Listy� Vol. 16, pp 256-264, 1922.
Available on the WWW.
- Die elektromotorische Wirksamkeit der Ionen, W. H. Nernst, �Zeitschrift f�r physikalische Chemie, St�chiometrie und Verwandtschaftslehre� Vol. 4, pp 129-181, 1889. Available on the WWW.
- Zur Kinetik der in L�sung befindlichen K�rper, W. H. Nernst, �Zeitschrift f�r physikalische Chemie, St�chiometrie und Verwandtschaftslehre� Vol. 2, pp 613-637, 1888. Available on the WWW.
- Experimental Researches in Electricity (2nd edition), M. Faraday, Richard and John Edward Taylor, London, 1849. Available on the WWW.
- The Bakerian Lecture: On Some New Phenomena of Chemical Changes Produced by Electricity, Particularly the Decomposition of the Fixed Alkalies, and the Exhibition of the New Substances Which Constitute Their Bases; And on the General Nature of Alkaline Bodies, H. Davy, �Philosophical Transactions of the Royal Society of London� Vol. 98, pp 1-44, 1808. Available on the WWW.
- Account of the new Electrical or Galvanic Apparatus of Alessandro Volta and Experiments performed with the same, W. Nicholson and A. Carlisle, �Journal of Natural Philosophy, Chemistry and the Arts� Vol. 4, pp 179-187, 1801. Available on the WWW.
- On the Electricity Excited by the Mere Contact of Conducting Substances of Different Kinds (in French), A. Volta, �Philosophical Transactions of the Royal Society of London� Vol. 90, pp 403-431, 1800. Available on the WWW, together with a partial English translation.
- Commentary on the effects of electricity on muscular motion (in Latin), A. Galvani, (Reprint of the original publication) �De Bononiensi Scientiarum et Artium Instituto atque Academia Commentarii� Vol. 7, pp 363-415, 1791. Available on the WWW, together with an English translation.
Listings of electrochemistry books, review chapters, proceedings volumes, and full text of some historical publications are also available in the Electrochemistry Science and Technology Information Resource (ESTIR). (http://knowledge.electrochem.org/estir/)
Return to:
Top –
Encyclopedia Home Page –
Table of Contents –
Author Index –
Subject Index –
Search –
Dictionary –
ESTIR Home Page –
ECS Home Page
|