Return to:
Encyclopedia Home Page –
Table of Contents –
Author Index –
Subject Index –
Search –
Dictionary –
ESTIR Home Page –
ECS Home Page
EXTRACTING METALS FROM SULFIDE ORES
Ronald Woods
School of Science, Griffith University
Nathan, Queensland 4111, Australia
E-mail: R.Woods@griffith.edu.au
(April, 2004; revised: October, 2010)
Many important metals are present in nature combined with sulfur as metal sulfides; these include cadmium, cobalt, copper, lead, molybdenum, nickel, silver, and zinc. In addition, gold and platinum group metals are associated with sulfidic base metal ores. The extraction of metals from their ores has been an essential factor in the creation and development of our advanced level of civilization.
Most sulfides are electrically conducting, generally semiconducting, and this allows electrochemical reactions to occur at the solid/solution interface In fact, electrochemical mechanisms play a key role in each of the processes that are involved in obtaining the component metals from their sulfide ores, namely, prospecting for ores (weathering of sulfide ore-bodies), separation and concentration of valuable components of ores (flotation), dissolving the valuable metal content of ores (extractive metallurgy), recovering metals from solution (electrowinning), and purifying the metal (electrorefining).
Weathering of sulfide ore-bodies
 |
| Fig. 1. Blasting sulfide ore in the Codelco Chuquicamata open-cut copper mine, Chile. |
The first step in recovering metals from their minerals is that of finding ore-bodies in which metal sulfides are present in a sufficient quantity, and at a sufficient concentration, for mining to be economic. One of the six basic geophysical exploration groups of methods commonly employed in the search for minerals is based on the sensing of mineral conductivity and of currents flowing as a result of oxidation of the sulfide minerals in natural weathering underground. The induced polarization exploration technique involves applying a series of current pulses to two electrodes inserted into the ground and analyzing the induced voltage at two different electrodes, also inserted into the ground. Electromagnetic measurements use alternating magnetic fields to induce measurable electrical currents in the earth and these depend on conductivity. This approach can be carried out from aircraft and it is then possible to screen large areas for later ground-based investigation. In the self-potential method, the potential between two electrodes inserted in the ground is measured and analyzed to detect electrochemical interaction between ore-bodies and ground water.
The electrochemical interaction between ore-bodies and ground water takes place by the same type of mechanism as occurs in the corrosion of metals. That is, the process involves anodic oxidation of the sulfide, a reaction that gives electrons to the mineral; these electrons are released again by the simultaneous cathodic reduction of oxygen. Since the sulfide ore-body is conducting, the anodic and cathodic processes can occur at different places. Thus, ore-bodies, when exposed to weathering processes, develop a zonal arrangement of different mineral associations, which reflect different degrees of oxidation. Oxygen dissolves in the ground water from the air and diffuses to metal sulfide surfaces where it can be reduced. This cathodic reaction takes up the electrons released by the anodic oxidation of sulfide minerals in the underlying ore-body and allows the process to proceed. Thus, electrons will flow through the ore-body to the surface and result in the ore at higher levels having a more positive potential than that at greater depth. The alteration of the composition of the ore-body will be greatest closer to the surface since the current path will be shorter. In this way, for example, alteration of ore-bodies containing the major economic copper mineral, a copper iron sulfide (chalcopyrite), often occur to form an overlying copper sulfide (chalcocite) zone and a cap containing oxidized copper minerals and native copper. It is the native copper that was the original source of this metal and was mined as early as 6,000 BC. Later copper mining involved leaching of copper oxides followed by cementation with iron. Cementacion is Spanish for precipitation, which better describes the process, and the use of the term cementation derives from the process being first carried out on an industrial scale in the copper mines in the Rio Tinto region of Spain in the mid 18th century.
Flotation separation
Valuable minerals in an ore can be separated from each other, and from worthless gangue minerals, by the froth flotation process. This process was developed in Australia at the start of the 20th century to treat the primary sulfidic silver/lead/zinc ore at Broken Hill, NSW. Some of the laboratory observations upon which the process depends were made during the nineteenth century in England, Germany, and the USA, but it was at Broken Hill that the first sustained efforts were made to exploit them, and that commercial scale operations became firmly established. The need for a new process to treat Broken Hill ore arose from the depletion of the secondary oxide ore that overlay the major sulfide mineralization. Natural oxidation to form the secondary zone had resulted in a concentration of silver and lead, and ore from this section of the ore-body could be smelted directly. The underlying sulfide zone, which comprised the bulk of the resource, was less amenable to treatment in the smelters of that era, and a method was required to recover the metal values if the potentiality of the primary ore was to be realized.
 |
| Fig. 2. Froth, loaded with sulfide mineral particles, flowing over the weir of a flotation cell. |
Many approaches were pursued to solve the "sulfide problem" at Broken Hill before selective flotation was developed. The successful technology involves first crushing the ore, typically to a particle size of about 5 to 50 micrometers, to liberate separate grains of the various valuable minerals and worthless gangue components. Then the particles are pulped with water, and the surface of the mineral of interest selectively made hydrophobic through the addition of an organic species, which is termed a collector. The term hydrophobic denotes "having little or no affinity for water" and means that the surface rejects water, as does an oily or waxy one. Following this procedure, a stream of air bubbles is passed through the pulp; the bubbles attach to, and levitate, the hydrophobic particles, which collect in a froth layer that disengages from the flotation cell by flowing over the weir of the cell. A frother, such as a long chain alkyl alcohol, is added to create a stable froth layer in the cell. The collector used at Broken Hill in the early days was eucalyptus oil derived from the leaves of the ubiquitous Australian "gum" tree. This led the President of the Mining Institute in Broken Hill at the time to proclaim, "Thus Nature, in close proximity to the vast bodies of complex ore, has provided the means for the concentration of such ores".
Following the success at Broken Hill, mining companies throughout the world rapidly adopted the flotation process. A similar situation to that at Broken Hill existed at the massive copper ore-body in Bingham Canyon in Utah, USA, where mining had exhausted the overlaying high-grade oxide ore by the end of late 19th century. The introduction of flotation in the early 20th century allowed the huge underlying low-grade copper sulfide mineralization to be processed economically.
A major advance in flotation practice was made in 1925 by the introduction of alkyl xanthates (dithiocarbonates) as collectors to replace compounds such as the eucalyptus oil initially used at Broken Hill. The xanthates proved to be much more selective than the compounds used previously and different sulfides could be sequentially floated by adjustments to the pulp chemistry. Xanthates are most efficient for the flotation of sulfides but quite inert for common gangue minerals. The flotation process has been continuously developed throughout the 20th century and into the 21st century to increase the efficiency of separation and to treat ever more complex ores. More effective and selective collector compounds than the xanthates have been developed and are used in practice, although xanthates are still applied widely. Most of these collectors are thiol compounds and derive their efficacy from chemical bonding between the metal atom in the sulfide and a sulfur atom in the collector.
The flotation cell contains an impeller to mix the pulp and inject air bubbles. The size of the cell has progressed over the years from a couple of cubic meters to up to 300 m3 today. Plants consist of multiple banks of cells and the process for each stage usually involves three steps. In the first group of cells, termed the rougher circuit, most of the mineral to be floated is recovered and this concentrate is then refloated in a cleaner circuit to make a more pure product. The sink fraction from the roughing stage is sent to a scavenger circuit and the subsequent float fraction is returned, together with the sink fraction from the cleaners, to the roughers to avoid losing valuable minerals.
 |
| Fig. 3. Wemco 160 cm3 cells in the flotation plant at Escondida, Chile, the biggest copper mine in the world. The impeller motor is seen on the top; the froth layer is inside the cell. (Courtesy of Minera Escondida.) |
Flotation is a key unit process in the recovery of most of the world copper, lead, molybdenum, nickel, platinum group elements, silver, and zinc, and in the treatment of certain gold and tin ores. And what is the electrochemical aspect of flotation? The interaction of the organic collector with the sulfide mineral surface occurs by a corrosion-type mechanism in which an anodic oxidation reaction attaches the organic collector to the mineral surface and the electrons transferred to the mineral by this reaction are returned to the solution phase by the reduction of oxygen. It thus follows the same type of mechanism as does ore oxidation discussed above.
Not only does the flotation process enable valuable sulfide minerals to be concentrated from worthless gangue minerals that make up the ore matrix, but it also allows different sulfide minerals to be separated from each other. For example, Broken Hill ore contains lead sulfide (galena), and zinc sulfide (sphalerite). In the first flotation stage, galena is rendered hydrophobic ("water rejecting") with an organic collector such as xanthate, while sphalerite is kept from floating by the addition of zinc sulfate, which deposits a hydrophilic ("water-loving"), wetting layer on its surface. Chemicals used to impede the flotation of a mineral, such as zinc sulfate in this case, are known in the industry as depressants. In the second flotation stage, copper sulfate is added and this compound is an activator. It reacts with the sphalerite surface to incorporate copper atoms and displace zinc atoms. Now the mineral surface can interact with collector molecules by the electrochemical process discussed above to make it hydrophobic and be incorporated into a froth layer, while leaving behind in the sink fraction oxides, carbonates, silicates, etc. Silver sulfide minerals in the ore follow the lead sulfide stream and cadmium sulfide follows the zinc. Silver and cadmium are separated from lead and zinc, respectively, in the subsequent metal extraction processes. The Broken Hill mines contain 6% lead and 10% zinc and flotation concentrates the ore into two streams, a lead concentrate containing 70% lead and a zinc concentrate containing 50% zinc. These concentrates are then sent to lead and zinc smelters, respectively.
Another example is the treatment of ores containing copper sulfide (chalcocite) and copper iron sulfide (chalcopyrite), together with molybdenum disulfide (molybdenite). The usual practice is to float both copper and molybdenum minerals in the first stage, leaving iron sulfides and other gangue minerals behind. Then the concentrate is sent to a second flotation circuit and sodium hydrosulfide added to depress the chalcopyrite by removing collector from its surface and the molybdenite floated alone, exploiting the fact that this mineral is naturally hydrophobic. Copper ores generally contain between 0.5% and 2% copper and the flotation process increases this to above 30%. The flotation concentrate contains sufficient copper for it to be suitable for smelting to recover the metal. Silver, gold and platinum group metals present in copper ores follow the copper in both the flotation circuit and the smelter, and are recovered in the final, electrorefining stage. Electrorefining is discussed in the final Section.
Flotation is also used to split copper and nickel sulfides and separate them from iron sulfides and gangue minerals. Cobalt sulfides appear in the nickel concentrate and the cobalt is subsequently recovered.
Flotation has developed from the treatment of simple ores to the ever more complex ones that are being found today. More selective collectors are used and the pulp chemistry adjusted to achieve optimum separations and recoveries. Thus, for example, the three metals in copper/lead/zinc ores can be floated into three concentrates, each containing one of the metals.
The process of flotation is sometimes also used in electrochemical waste treatment .
Extractive metallurgy
Metal sulfide flotation concentrates can be treated in a number of ways. These include using  pyrometallurgy, or smelting at high temperature, in which process the sulfur in the metal sulfide is oxidized with air or oxygen to sulfur dioxide, and molten metal is produced. The sulfur dioxide needs to be captured for air-quality reasons and is converted into sulfuric acid. Treatment of sulfide mineral flotation concentrates can also be carried out using hydrometallurgy, in which process the metal sulfide is dissolved (leached) into an aqueous solution. Some processes involve both smelting and leaching steps. This is the case, for example, in zinc sulfide processing. The zinc concentrate is first roasted to release sulfur dioxide and form zinc oxide and then the latter compound is dissolved in sulfuric acid and the metal extracted by electrolysis. The recovery of zinc by electrolysis is presented in the Electrowinning Section. pyrometallurgy, or smelting at high temperature, in which process the sulfur in the metal sulfide is oxidized with air or oxygen to sulfur dioxide, and molten metal is produced. The sulfur dioxide needs to be captured for air-quality reasons and is converted into sulfuric acid. Treatment of sulfide mineral flotation concentrates can also be carried out using hydrometallurgy, in which process the metal sulfide is dissolved (leached) into an aqueous solution. Some processes involve both smelting and leaching steps. This is the case, for example, in zinc sulfide processing. The zinc concentrate is first roasted to release sulfur dioxide and form zinc oxide and then the latter compound is dissolved in sulfuric acid and the metal extracted by electrolysis. The recovery of zinc by electrolysis is presented in the Electrowinning Section.
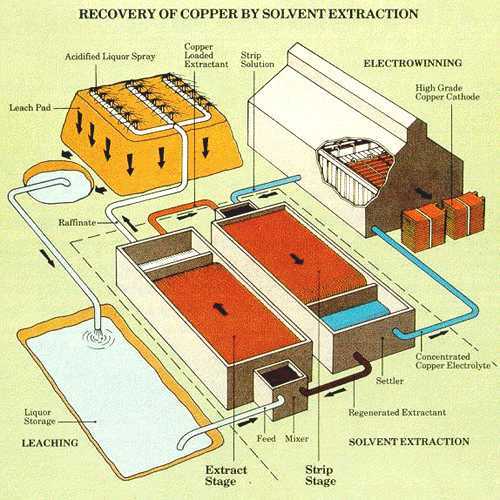
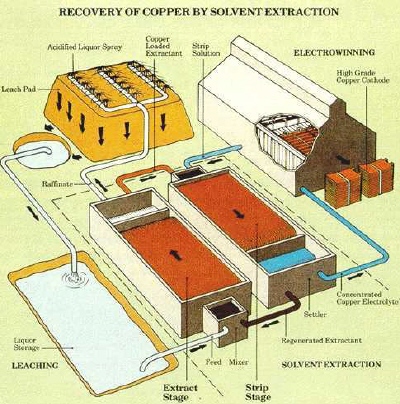 |
| Fig. 4. Illustration of the SX/EW process for recovering copper by heap leaching. (Courtesy of Cytec Industries, suppliers of chemicals to the minerals industry.) |
Environmental restrictions on sulfur gas emissions in smelting operations, and the limited market for sulfuric acid, resulted in a search for hydrometallurgical routes from sulfide minerals to metals. In the 1950s, the Sherritt-Gordon ammonia pressure-leaching process for nickel was introduced. In this process, nickel sulfide flotation concentrates are reacted under pressure with oxygen together with ammonia, which helps to facilitate the dissolution by forming soluble nickel-ammonia complex ions. As with flotation, dissolution is a corrosion-type process in which anodic oxidation of the sulfide transfers electrons to the remaining mineral and these electrons are returned to the solution by the cathodic reduction of oxygen. Nickel is recovered from the solution in the Sherritt-Gordon process either by electrolysis or by reduction with hydrogen gas. The latter process, too, is an electrochemical one, and needs the addition of fine nickel particles as seeds. The hydrogen is anodically oxidized to hydrogen ions on the nickel seed and gives electrons to the metal. These electrons are used up in the cathodic reduction of nickel ions to form nickel metal, which deposits on the seed particles. The nickel particles are removed when they reach a suitable size.
The success of the nickel hydrometallurgical process led to research for similar methods to recover other metals. The Sherritt-Gordon pressure leach procedure for the treatment of zinc sulfide concentrates was introduced in 1981; zinc sulfide is oxidized by oxygen in sulfate solutions to form zinc sulfate and sulfur. The zinc is recovered by electrolysis as it is in the roast leach process already described.
Leaching of copper from ores has focused mainly on oxide ore types. Most oxide copper ores are not amenable to present-day flotation techniques and are not rich enough for direct smelting. Thus, the winning of copper from these ores must involve hydrometallurgical techniques. Most oxidized copper minerals dissolve readily in sulfuric acid so that leaching is reasonably straightforward. A concentrated, pure copper sulfate solution suitable for electrowinning is produced from the initial leach solution by selectively transferring the copper ions to an organic phase by a process known as solvent extraction and returning them again to an aqueous phase. Complexing agents are dissolved in an organic solvent such as low vapor pressure kerosene and this phase is intimately dispersed within the aqueous leach solution in a mixer/settler (see Figure 4). The copper selectively complexes with the reagent and is transferred into the organic phase. The copper replaces hydrogen on the complex and hydrogen ions are consequently transferred to the aqueous phase. The two phases are allowed to separate in the settler section and then the aqueous acid solution, termed the raffinate, is recycled to ore leaching. The loaded organic phase is sent to a second mixer/settler where it is reacted with a strong aqueous sulfuric acid solution. Here, the copper is exchanged for hydrogen ions and transferred into the second aqueous phase from which the metal is efficiently recovered by electrolysis. The overall process of dissolution, solvent extraction and electrowinning is known as SX/EW. Copper electrowinning is discussed in the Electrowinning Section.
Mixed oxide and sulfide copper ores are leached following the same process stages as in Figure 4. Also, some sulfidic ores that are too lean to treat by flotation are also leached in heaps or dumps. The recovery of metals from sulfides in this way utilizes bacteria either naturally present or inoculated into the material. The microorganisms aid the oxidation of insoluble metal sulfides to soluble metal sulfates. The commercial application of bacteria to accelerate sulfide leaching in a controlled manner is a relatively new concept. Bacteria did not, of course, wait for man to exploit their activity. The presence of copper in mine drainage reported in early Roman times was, no doubt, a result of bacterial leaching. The treatment of mine waters to recover dissolved copper was applied as early as 1670 at Rio Tinto in Spain and in the 19th century in the USA.
Into the future
The rate of the leaching of sulfides in sulfuric acid media is significantly less than that of oxides, but the intervention of bacteria accelerates the process. Bacterial leaching is being developed to operate much more effectively than that occurring naturally. Effective bacteria in sulfide leaching include Thiobacillus ferrooxidans, Thiobacillus thiooxidans, and Leptospirillum ferrooxidans. Mixed cultures of these bacteria solubilize metal sulfides more effectively than do pure cultures (that is, cultures containing only one species of bacteria).
The bacteria do not alter the electrochemistry of the leaching process, but rather catalyze individual reactions. For example, oxygen passes through the cell of the microorganism and is reduced in the cell matrix. The electrons released in this cathodic process are transferred via the cell cytochromes and through the cell wall to oxidize double- charged iron cations in solution to triple-charged iron cations. The triple-charged iron then chemically attacks the metal sulfides and converts them to dissolved metal ions. The hydrogen ions consumed by oxygen reduction maintain a high pH in the intercellular space to allow the bacteria to function while residing in a strong acid environment (pH 1-3). The electron transfer reactions involving iron and oxygen provide the bacterium with the energy required for life. The action of the bacteria maintain the triple-charged iron concentration in solution at a higher level than that existing under sterile conditions and hence the rate of leaching of sulfides is enhanced. The above-mentioned bacteria also oxidize sulfur to sulfate and this can also accelerate leaching through the removal of inhibiting surface layers. Thus, insoluble metal sulfides are oxidized to soluble metal sulfates.
Biochemical leaching is now a well-established commercial process for treating gold ores in which the gold is enclosed within sulfide minerals. Biochemical oxidation of the sulfide minerals releases the gold and allows it to be leached by the chemicals used in conventional gold leaching.
Biochemical processes are being developed for the treatment of copper, nickel and cobalt sulfide ores and are expected to become the major method for recovery of these metals in the future. The aim is to eventually replace smelting processes that generate sulfur dioxide.
The ultimate advance in recovering metals from sulfide ores would be to eliminate the mining stage and leach the ore-body where it is in the ground. This would involve pumping a leach solution underground and pumping up the reacted solution containing dissolved valuable metal species. Such in-place leaching has already been practiced commercially in uranium and copper mines. The best results have been obtained in the recovery of metals from residual ore in mines in which the high-grade ore has been mined out since mine workings provide access of the solution to the ore. The major problem with new ore-bodies is in breaking the ore underground to make it accessible to the leach solution without having physical access to it.
Electrowinning
Metals can be recovered from solution by electrolysis, a process that is known in the extractive metallurgy industry as electrowinning. When two electrodes are placed in a solution containing metal ions and an electric current is passed between them, the metal can be deposited on the negative electrode. In the recovery of most metals, oxygen is evolved from water at the positive electrode. An electrolyte, and a current density, is generally chosen that gives a dense, compact electrodeposit, and additives included in the electrolyte to further improve product quality (a practice also used in electroplating).
Large-scale electrowinning of copper was developed between 1912 and 1915 at one of the world largest copper mines, that at Chuquicamata in Chile. Electrowinning has become the exclusive choice for recovering copper following leaching of oxide or sulfide ores and solvent extraction. SX/EW produced more than 3.3 million tonnes of copper in 2009, which is 21% of total world copper production.
In a typical electrowinning or electrorefining operation, the electrolytic cells are rectangular tanks and each contains 20-50 negative electrodes and a similar number of positives. An additional positive electrode is required in each cell in order to ensure plating occurs on both sides of each negative. The electrodes are designed to rest on busbars that supply electricity; these are situated outside the top of each tank, one for the negatives and another for the positives. Thus, the electrode pairs in each tank operate in parallel. In the tankhouse, there is a multiple system of practical sections with the banks of cells connected in series and parallel to obtain optimum use of the electrical power derived from the rectifiers, while keeping the voltage to earth at any point at a level that does not pose a risk to personnel.
In copper electrowinning, the positive, oxygen evolving, electrodes are usually made from lead calcium alloys similar to those used for the grids in lead-acid batteries. The traditional negative electrodes for copper electrowinning were thin copper starter sheets made by plating copper onto titanium or stainless steel electrodes. The copper was then stripped from the substrate and inserted into the electrolysis cell and copper deposited from a solution usually containing a concentration of 25 to 60 grams/liter (g/l) copper as copper sulfate and 50 to 180 g/l sulfuric acid, held at 50 to 60oC (122-140oF). Small quantities of a hydrocolloid such as Guar gum are included in the electrolyte, as this helps to form a dense electrodeposit. Typically, a current equivalent to a current density of 300 A/m2 is passed between each pair of positive and negative electrodes; this gives a cell voltage of ~2.0 V and the energy consumption is ~2.0 kWh/kg.
In an electrolysis cell such as in electrowinning or refining, the negative electrode is the cathode and the positive electrode is the anode. Hence electrowon, or electrorefined, copper is known as cathode copper.
Modern developments in copper electrowinning include the use of re-usable stainless steel electrodes to replace copper starter sheets. This technology was established in the Isa Process and later in the Kydd Creek Process. In addition to lowering manpower requirements, it allows the current density to be increased by ~10%. Following electrolysis, the copper is stripped from the stainless steel in special automated machines.
In a typical commercial copper electrowinning plant, there will be thousands of pairs of electrodes in operation together. In modern systems, insertion of negative electrode blanks, removal of loaded electrodes, and transfer to the stripping machines are fully automated.
 |
| Fig. 5. Zinc electrowinning at the Teck Cominco zinc refinery in Trail, BC, Canada. The top of the cells are shown revealing the multiplicity of electrodes. Above the cells, loaded electrodes are being harvested. (©Teck Cominco Metals Ltd. Reproduced with permission.) |
As pointed out above, zinc electrowinning from zinc sulfate solution is the usual final stage in the recovery of zinc from sulfide flotation concentrates. Electrowinning was introduced in the second decade of the twentieth century to replace distillation. As with copper, zinc is deposited on the negative electrode and oxygen is evolved, derived from water, at the positive electrode. The electrolyte typically contains 55 to 70 grams/liter of zinc as zinc sulfate and 150 to 200 g/l of sulfuric acid and is held at 35 to 38oC (95-100oF). Small, controlled quantities of glue and antimony are added to the electrolyte to help form a smooth deposit. A current density between 400 and 800 A/m2 is employed, and this gives a cell voltage of ~3.5 V and an electrical energy consumption of ~3.3 kWh/kg. The overall reaction converts aqueous zinc sulfate to zinc metal and sulfuric acid. The acid is then recycled to the leaching stage where zinc oxide is dissolved. The positive electrodes in zinc electrowinning are made from lead silver alloys; oxygen evolution is promoted on these alloys and lead dissolution, which would contaminate the zinc product, is avoided. More than 80% of the 2009 world zinc production of 11 million tonnes was produced by electrowinning.
Zinc is, in principle, more difficult to electrowin than copper because zinc lies considerably above hydrogen in the electrochemical series, whereas copper lies below. Thus, the evolution of hydrogen from an acidic zinc sulfate solution is energetically favored over zinc deposition. As it happens, hydrogen evolution on a zinc surface is a very slow process (it occurs at a high overpotential) and hence zinc can be electrodeposited from acid solutions. But impurity metal ions in the solution can plate out as well, if they are below zinc in the electrochemical series. These include antimony, arsenic, cobalt, copper, germanium, and nickel. Not only would these elements contaminate the zinc product if they are present in significant quantities, but also they can promote cathodic hydrogen evolution on the negative electrodes and hence diminish the current efficiency of electrolysis to deposit zinc. This means that the solution needs to be purified before electrolysis. Purification of the solution is achieved by adding zinc dust. Zinc will displace elements below it in the electrochemical series and remove the impurities that effect electrowinning efficiency. These cementation, or metal displacement, reactions also take place by an electrochemical process. The anodic dissolution of zinc gives up electrons to the zinc particles and these are used up in the coupled cathodic reactions, which are the deposition (and removal) of the impurity elements from the solution.
Cadmium metal produced in the purification of zinc electrowinning electrolyte is dissolved in sulfuric acid solution and electrowon in a process similar to that for zinc.
About 60% of the world 2009 nickel production of 1.4 million tonnes was derived from sulfide ores, but an ever-increasing proportion is coming from other ores (such as laterites, in which nickel is present in an oxy-hydroxide or silicate matrix). Indeed, over 70% of the world reserves of nickel are lateritic. Laterites are usually leached to dissolve the nickel. The recovery of nickel from solution, following leaching of either type of nickel ore, can be achieved by hydrogen reduction, as discussed above, or by electrolysis. Outokumpu uses the latter technology in Finland. Nickel is deposited from nickel sulfate solutions (~60 grams/liter nickel) containing sodium sulfate and boric acid to improve the deposit. The latter additive adjusts the pH to ~3.5. The metal is deposited at 65oC (149oF) onto nickel starter sheets and oxygen is evolved at the lead positive electrodes. Nickel is also electrowon in a form suitable for use as soluble positive electrodes in nickel electroplating.
About 70% of the cobalt world 2009 production of 62,000 tonnes was also recovered by electrolysis in a process similar to that for nickel.
Electrorefining
 |
| Fig. 6. The Xstrata copper electrorefining tankhouse at Copper Refineries in Townsville, Queensland, Australia, which developed and uses the ISA PROCESS technology. The picture shows the top of the cells and the multiplicity of electrodes. The negatives are the most visible; between each can be seen the lugs at the ends of the positives. The crane manipulating the electrodes is automated. (Courtesy of Xstrata Copper.) |
Electrorefining is an electrolytic process that involves anodically dissolving a metal at the positive electrode in a cell and simultaneously re-depositing the same metal at the negative electrode. The first patents for metal electrorefining were for copper and were granted in 1865. The first successful plant was built in Pembrey, Wales in 1869 and this was followed in 1876 by the Norddeutsche plant in Hamburg, Germany, which is still operating today and is the largest copper refinery in Europe. The first commercial operation in the USA was constructed in Newark, New Jersey in 1883. The Newark plant produced 2-3 tonnes of electrolytic copper per day during its first year. This contrasts with the electrorefining capacity at Chuquicamata in Chile in 2009 of over 2,300 tonnes per day.
Electrorefining is the final step in the smelting process for the recovery of copper from sulfide ores, and this process accounted for nearly 80% of the world copper production of 16 million tones in 2009. Smelting produces blister copper, which contains about 99% copper and this is electrorefined to cathode copper that has a purity of around 99.99% copper. The positive electrodes for the electrorefining cell are cast from the blister produced by smelting copper sulfide flotation concentrates, and weigh 300 to 380 kg; the negative electrodes are the same as in copper electrowinning discussed in the previous section. The electrolyte contains 40 grams/liter of copper as copper sulfate and 150 to 200 g/l sulfuric acid and the refining cells operate at ~60oC (~140oF). Small quantities of glue, thiourea and chloride are added to the electrolyte; these compounds adsorb on the copper surface and help to form a dense electrodeposit. A current density of ~200 A/m2 is passed between the negative and positive electrodes and this results in a cell voltage of about 0.28 V. The electrical energy consumption in the copper electrorefining cell is ~0.25 kWh/kg.
Electrorefining is successful because metallic impurities that are more noble than copper, that is, are lower in the electrochemical series, do not enter the electrolyte when copper dissolves at the positive electrode. These impurities include precious metals – silver, gold and the platinum group metals – which are recovered by treatment of the sludge that falls to the bottom of the cell. Since impurities that do enter the electrolyte are less noble than copper, that is, are above copper in the electrochemical series, do not deposit on the negative electrode. Some metals, for example, nickel, accumulate in the electrolyte and are extracted in a separate stage.
Electrorefining of copper is a large operation. For example, an average copper refinery producing 500 tonnes of metal per day needs 0.2 km2 (kilometer = one thousand meters) of total electrode area. This corresponds to about 50,000 positive and 50,000 negative electrodes suspended in about 1,500 tanks occupying a total floor area of about 6,000 m2.
 |
| Fig. 7. Lead electrorefining at the Teck Cominco smelter in Trail, BC, Canada. (©Teck Cominco Metals Ltd. Reproduced with permission.) |
Lead is also purified by electrorefining using the Betts process pioneered by Cominco at its smelter in Trail, BC, Canada in 1902. Lead from the smelter is melted and cast into slabs, which act as positive electrodes. Refined lead is cast into thin starter sheets for the negative electrodes on which lead is deposited. The electrolyte used is ~90 grams/liter hydrofluorosilicic acid containing ~70 g/l lead as lead hydrofluorosilicate and operates at 30 to 40oC (86-104oF). Glue and lignin sulfonate are included in the electrolyte to help form a smooth deposit. A current density of between 160 and 200 A/m2 is used and this gives a cell voltage of 0.35 to 0.65 V. The energy consumption is ~0.13 kWh/kg. Refining of lead is also carried out in sulfamide media. In 2009 the world production of lead was 4.0 million tonnes from mined ore and a further 4.8 million tonnes from secondary (recycled) sources. About 12 % of the world�s production of lead is electrorefined.
Nickel may also be purified by electrorefining; this process competes with the carbonyl process that exploits the volatility of nickel carbonyl.
Silver refining, which is carried out following the separation of this metal in copper and lead smelting, is especially interesting. While the aim in other metal electrorefining processes is to produce a massive, dense product, with silver, fine, tree-like dendrites are formed deliberately. This is achieved by plating the metal onto a substrate such as stainless steel or carbon from an electrolyte that does not complex with silver, plating from a quiescent electrolyte without the inclusion of additives that promote smooth deposits, and operating at a high current density. The silver dendrites formed on the negative electrodes can be easily harvested since they do not attach strongly to the electrode surface. The electrolyte typically contains 150 g/l of silver as silver nitrate and nitric acid to make the pH 1-1.5. The cell operates at ~35oC (~95oF) and a current density is ~400 A/m2. The positive electrodes are cast silver derived from the slimes formed in electrorefining of copper or lead. The electrorefined metal contains between 99.9% and 99.99% silver. The anodic slimes from silver refining are further treated to recover gold and platinum group metals.
Acknowledgement
This article was, by permission, translated into Ukrainian and posted at:
http://webhostinggeeks.com/science/m02-metals-ua
This article was, by permission, translated into Romanian and posted at:
http://webhostinggeeks.com/science/electrochemistry-encyclopedia-rm
Related articles
Aluminum production
Current density distribution in electrochemical cells
Electroplating
Metal powder production by electrolysis
For other industrial electrolytic processes see:
Brine electrolysis
Industrial organics
Bibliography
- Electrochemistry in mineral and metal processing 8 (Proceedings of the 8th International Symposium, From the 217th Meeting of The Electrochemical Society, Vancouver, Canada, April 26-28, 2010, ECS Transactions, Vol. 28, No. 6), F.M. Doyle, R. Woods, and G.H. Kelsall (editors), ECS, Pennington, NJ 2010.
- Electrochemistry in mineral and metal processing VII (Proceedings of the 7th International Symposium, From the 209th Meeting of The Electrochemical Society, Denver, Colorado, May 8-10, 2006, ECS Transactions, Vol. 2, No. 2), F.M. Doyle, G.H. Kelsall, and R. Woods (editors), ECS, Pennington, NJ 2006.
- Electrochemistry in mineral and metal processing VI (Proceedings of the 6th International Symposium, From the 203rd Meeting of
The Electrochemical Society, Paris, France, April 27-May 2, 2003, ECS
Proceedings, Vol. 2003-18), F.M. Doyle, G. H. Kelsall, and R. Woods (editors), ECS, Pennington, NJ 2003.
- Hydrometallurgy 2003 – Fifth International Conference in Honor of Professor Ian Ritchie (two volumes), C. Young, A. Alfantazi, C. Anderson, A. James, D. Dreisinger, and B. Harris (editors), The Minerals Metals and Materials Society, Warrendale, PA, 2003.
- Electrochemistry in mineral and metal processing V (Proceedings of the 5th International Symposium, From the 197th Meeting of The Electrochemical Society, Toronto, Canada, May 14-18, 2000, ECS Proceedings, Vol. 2000-14), R. Woods and F.M. Doyle (editors), ECS, Pennington, NJ 2000.
- Electrochemistry in mineral and metal processing IV (Proceedings of the 4th International Symposium, From the 189th Meeting of The Electrochemical Society, Los Angeles, California, May 5-10, 1996, ECS Proceedings, Vol. 96-6), R. Woods, F.M. Doyle, and P. Richardson (editors), ECS, Pennington, NJ 1996.
- Industrial Electrochemistry (2nd edition), D. Pletcher and F.C. Walsh, Kluwer, Dordreck, 1993.
- Electrochemistry in mineral and metal processing III (Proceedings of the 3rd International Symposium, From the 181st Meeting of The Electrochemical Society, St. Louis, Missouri, May 17-21, 1992, ECS Proceedings, Vol. 92-17), R. Woods and P.E. Richardson (editors), ECS, Pennington, NJ 1992.
- Electrochemistry in mineral and metal processing II (Proceedings of the 2nd International Symposium, From the 173rd Meeting of The Electrochemical Society, Atlanta, Georgia, May, 1988, ECS Proceedings, Vol. 88-21), P.E. Richardson and R. Woods (editors), ECS, Pennington, NJ 1988.
- Electrochemistry in mineral and metal processing (Proceedings of the 1st International Symposium, From the 165th Meeting of The Electrochemical Society, Cincinnati, Ohio, May, 1984, ECS Proceedings, Vol. 84-10), P.E. Richardson, S. Srinivasan, and R. Woods (editors), ECS, Pennington, NJ 1984.
- Comprehensive Treatise on Electrochemistry, Vol. 2: Electrochemical Processing, J.O'M. Bockris, B.E. Conway, E. Yeager and R E. White (editors), Plenum, NY, 1981.
Listings of electrochemistry books, review chapters, proceedings volumes, and full text of some historical publications are also available in the Electrochemistry Science and Technology Information Resource (ESTIR). (http://knowledge.electrochem.org/estir/)
Return to:
Top –
Encyclopedia Home Page –
Table of Contents –
Author Index –
Subject Index –
Search –
Dictionary –
ESTIR Home Page –
ECS Home Page
|